info@biomedres.us
+1 (502) 904-2126
One Westbrook Corporate Center, Suite 300, Westchester, IL 60154, USA
Site Map
Received: May 06, 2026; Published: May 19, 2026
*Corresponding author: Alber Fares, Professor of Medical Biochemistry and Genetics, Orlando, Florida, USA
DOI: 10.26717/BJSTR.2026.65.010232
Enzymes serve as biological catalysts and are essential to numerous biochemical processes; significantly advancing biotechnology. Enzyme kinetics plays a critical role across disciplines such as enzymology; medicine; biocatalysis; and metabolic engineering. This study delves deeply into enzyme kinetics and catalytic mechanisms; aiming to enhance their application across different biotechnological fields. The research offers valuable insights into modulating enzyme activity; paving the way for the development of more efficient and resilient biocatalysts. These innovations are crucial for drug discovery; industrial biocatalysis; and mechanistic enzymology; underscoring the importance of enzymes across a range of scientific and practical areas. Furthermore; this study aims to aid researchers in accurately interpreting and reporting enzyme behavior; thereby bridging gaps in interdisciplinary research. A solid understanding of enzyme dynamics has profound implications for designing enzymes tailored to meet specific industrial requirements; ultimately boosting the efficiency and sustainability of biotechnological processes.
Keywords: Biological Catalysts; Biochemical Reactions; Enzyme Kinetics; Catalytic Mechanisms; Catalytic Efficiency; Enzyme Kinetics; Enzymology; Michaelis–Menten kinetics; Transient-State Kinetics; Steady-State Kinetics; Rapid-Equilibrium Kinetics
Abbreviations: MM equation: Michaelis-Menten Equation; ES: Substrate Complex; k2: Specific Constant Rate; E: Enzyme; ES: Enzyme-Substrate Complex; S: Substrate; P: Product; V0: Initial Velocity; Vmax: Maximum Velocity; Km: Michaelis-Menten Constant; kcat: Turnover Number (the Maximum Number of Product Molecules Generated Per Enzyme Molecule Per Unit of Time); Kᵢ: Inhibition Constant
Enzymes are complex protein molecules produced by living cells that act as biocatalysts [1]. They are highly specific; typically catalyzing a single chemical reaction or closely related reactions; and minimizing side reactions that produce unwanted by-products compared to uncatalyzed reactions [2]. The interaction between an enzyme and its substrate is a crucial event in enzyme catalysis; significantly influencing the specificity and efficiency of biochemical reactions. Enzymes are typically globular proteins; and their unique three-dimensional structure leads to the formation of an active site—a specialized region designed for binding substrate molecules through various non-covalent interactions. These interactions include hydrogen bonding; ionic interactions; hydrophobic forces; and van der Waals interactions; all of which contribute to the formation of a transient enzyme-substrate complex (ES) [3].
Enzyme kinetics is essential in systems biology for understanding biological networks. Mathematical models of enzyme kinetics reveal principles governing biochemical reactions; with enzyme activity influenced by substrate concentration; availability; and environmental factors. While traditional experiments offer insights; they often fall short in capturing complex interactions and predicting behavior across different conditions. Recently; researchers have focused on the dynamics of fractional-order models [4]. Assessing an enzyme’s catalytic activity is crucial for its comprehensive characterization. While the specific methodology tailored for a given enzyme is influenced by various factors; it is ideal to implement a technique that enables continuous observation of either substrate depletion or product formation— typically represented as an absorbance-versus-time graph. This data allows for the calculation of the initial velocity; defined as the initial slope of the reaction under specified conditions [5]. [6] Enzyme kinetics studies the rates of enzymatic reactions; the factors influencing these rates; and how enzymes interact with substrates. It is essential in biochemistry and medicine; providing a framework for analyzing enzyme behavior. Kinetic analysis assesses enzyme efficiency; substrate affinity; and regulatory control; linking molecular interactions to metabolic functions [7]. At the molecular level; enzymatic reactions involve intermediate steps. Transition-state theory states that enzymes enhance reaction rates by stabilizing the high-energy transition state; lowering the activation energy without altering the overall free energy change. Understanding transition-state formation is vital for interpreting kinetic parameters and designing enzyme inhibitors that mimic the transition-state structure of substrates [8,9].
Michaelis and Menten (1913) highlighted that enzyme activity is highly dependent on specific conditions; such as temperature; pH; and ion strength. While it seems straightforward to establish general rules for enzyme assays; the diversity among enzymes makes this difficult. Enzymes function best at their optimum conditions; and deviations can reduce activity. Moderate deviations may lead to tolerable decreases in activity; which align with physiological conditions in mammalian cells. However; assay procedures are tailored to individual enzymes rather than general standards. Additionally; some enzymes; especially those from extremophiles; require conditions outside the physiological range to be active [10]. Temperature fluctuations play a crucial role in modulating substrate diffusion and enzyme kinetics within protein crystals. This alteration enhances enzyme turnover and facilitates a more thorough characterization of enzymatic mechanisms. Consequently; multidimensional experiments can significantly advance ongoing contributions in structural biology; leading to a deeper understanding of conformational dynamics and their influence on protein function [11].
Enzyme inhibition is crucial in enzyme kinetics and metabolic regulation. Inhibitors can reduce enzyme activity through various interactions: competitive; non-competitive; uncompetitive; or mixed. Mixed inhibition affects both substrate binding and catalytic activity. Kinetic analysis reveals insights into enzyme active sites and drug action. Additionally; enzymes can be regulated by substrate and product inhibition; where high substrate concentrations or reaction products modulate activity. Many regulatory enzymes do not follow classical Michaelis-Menten kinetics; they may exhibit cooperative behavior and allosteric sites that; when bound by effectors; alter activity and impact metabolic flux. Understanding these dynamics is vital for grasping complex metabolic regulation [11]. Assessing catalytic rates and binding affinities of substrates and inhibitors is essential for characterizing enzymes and evaluating their applications; including inhibitor drug development; biocatalytic synthesis; understanding drug elimination; and revealing enzyme reaction mechanisms [12].
The Michaelis-Menten equation is highly regarded for its ability to predict enzyme behavior; particularly in substrate binding and catalysis; even before the role of enzymes as proteins was fully understood. Originally based on studies of invertase; the equation has broad applicability to most enzymes. This guide explores its origins; mathematical significance; and the extensive research leading to this expression. It also examines advancements beyond the steady-state assumption; including insights from pre-steady-state; single turnover; and full progress curve modeling in enzyme kinetics [13]. We can categorize kinetic analyses of enzymes and their corresponding parameters into three main groups:
1. Transient-state (pre-steady-state) kinetics;
2. Steady-state kinetics; and
3. Rapid-equilibrium kinetics [14-18].
Modifiers of enzyme-catalyzed reactions significantly influence reaction velocity [19,20]. When a single molecule of a modifier binds to an enzyme; it alters the kinetic mechanism’s determinants; yielding two rate constants. Conversely; if two modifier molecules engage in two separate reactions; there will be five independent equilibriums and three pathways for product synthesis [20]. Under laboratory conditions; factors such as drugs; toxins; radicals; activators; heavy metals; and pH can significantly affect the attainment of a chemical equilibrium. However; the kinetic behavior of enzymes varies considerably under cellular conditions due to a multitude of influencing variables [21]. When enzymes malfunction; it can lead to serious diseases [22,23]. For over a century; the classical approach to understanding enzyme kinetics has relied on the Michaelis-Menten equation (MM equation); originally formulated by Michaelis and Menten (Scheme 1) [24]. Kinetic models serve as valuable tools for optimizing biocatalytic reactions; aiding process design and scaling to enhance productivity while lowering costs across various processes [25,26]. Kinetic studies in enzymology can be categorized into three distinct types: transient-state kinetics; steady-state kinetics; and rapid-equilibrium kinetics [27].
o Transient-state kinetics focuses on extremely rapid reactions; where the mechanisms are closely linked to the enzyme’s structure [19,28].
o Steady-state enzyme kinetics operate under the premise that the steps in the catalytic process maintain a constant state; even when subjected to continuous changes [29].
o Rapid-equilibrium kinetics involves reactions that reach equilibrium with their components—such as the enzyme; substrate; and enzyme-substrate complex—prior to the rate-determining step [20].
Steady-State Kinetics
The Michaelis–Menten equation was initially derived under the assumption that substrate binding occurs at equilibrium (Scheme 1) [30]. The initial collision of enzyme (E) and substrate (S) leads to a bimolecular reaction with a second-order rate constant (k1). The enzyme substrate complex (ES) can then convert to enzyme (E) and product (P) (first-order rate constant k2) or revert to E and S (first-order rate constant k-1). The reverse formation of ES from E and P is not depicted; as it is assumed that product formation from ES (k2) does not reverse [31].
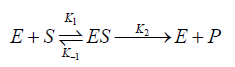
Scheme 1: Michaelis–Menten simplest form [31].
To determine the rate constant; k2; for a reaction; one must know the reaction velocity and the concentration of the enzyme-substrate complex (ES). The reaction velocity can be easily measured using a substrate that produces a detectable product. However; measuring ES is more challenging; as its concentration relies on both the formation and decomposition rates of the enzyme-substrate complex [31] (see Figure 1).
Figure 1

Later; the steady-state approximation was applied to incorporate the rates of substrate and product release; thereby defining Km (Michaelis constant) within a simpler model (Scheme 2). This model provides a foundational framework for analyzing enzyme kinetics by relating the reaction rate to substrate concentration [30].
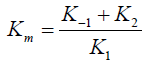
Scheme 2: Michaelis–Menten form [30].
The Michaelis constant Km represents the substrate concentration at which the reaction rate is half of its maximum velocity Vmax. Understanding Km is crucial; as it offers insights into the enzyme's affinity for the substrate—lower Km values indicate higher affinity [32]. Another important parameter is Vmax; which reflects the maximum reaction rate when the enzyme is saturated with substrate. Together; Km and Vmax allow researchers to characterize enzyme behavior under various conditions; aiding comparisons of different enzymes or the same enzyme under different environmental conditions [33] (see Figure 2). The Km indicates the substrate concentration at which the initial reaction velocity V0 is half of Vmax; reflecting the enzyme's affinity for its substrate; lower Km signifies stronger ES interactions. Typically expressed as Km = (k-1 + k2)/k1 (Scheme 2) Michaelis–Menten form; it can become complex in reactions with multiple steps. When k-1 is much greater than k2 (k-1 > k2); Km approximates the dissociation constant Kd (k-1 + k2) [30,31]
Figure 2

These kinetic parameters are invaluable in biochemistry and pharmacology; as they guide the design of drugs and inhibitors by predicting how changes in enzyme concentration or environmental conditions might affect reaction rates. By manipulating these conditions; scientists can optimize enzyme efficiency for industrial applications or develop targeted therapies that modulate enzyme activity in disease states [34].
Steady-state kinetics analysis measures initial rates (V0) at a constant enzyme concentration while varying substrate concentration; usually with substrate levels significantly higher than enzyme concentration [35-37]. In the initial reaction phase; the formation rate of the ES complex (Michaelis complex) is almost equal to its breakdown rate; indicating steady-state conditions. This is achieved by using a small fraction of the substrate (about 10% or less) during initial velocity measurements; keeping the substrate concentration nearly constant; which results in pseudo-first-order kinetics for the first step in Scheme 3 [38]. The enzyme kinetics model involves a substrate binding to an enzyme; with rates of association k1 and dissociation k2; followed by catalysis kcat to produce a product and regenerate the enzyme. Enzymes lower the energy barrier between substrate and product and can modify single or multiple molecules. The Michaelis- Menten model describes enzyme reactions; where S is substrate concentration; Vmax is the maximum reaction rate; and Km indicates the reaction’s sensitivity to substrate [39].
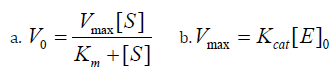
Scheme 3: (a.) The initial rates (b.) the relationship between Vmax and kcat [36].
[40] This content discusses key kinetic metrics from steady-state kinetic analysis; focusing on Vmax and kcat.
1. Vmax indicates the maximum enzyme rate at saturating substrate
concentration and should be reported with enzyme
concentration for context.
2. kcat; the turnover number; shows the maximum product molecules
generated per enzyme per time and is independent of
concentration.
3. kcat and kcat/Km allow for comparisons across different enzymes
under similar conditions.
4. kcat is usually equivalent to k2 when k-1 > k2.
Vmax /Km or kcat/Km; known as the “specificity constant” or “catalytic efficiency;” measures an enzyme's substrate preference; facilitating comparisons among enzymes or substrates. Literature emphasizes kcat and kcat/Km as key kinetic parameters from steady-state analysis; while Km is seen as a ratio. The value kcat/Km represents the apparent second-order rate constant for the substrate's productive binding to the enzyme; closely aligning with a true second-order rate constant when the formation of the product occurs faster than the reverse reaction [18,30,40,41]. Calculating Vmax /Km or kcat/Km can be achieved through several methods. One approach involves applying non-linear regression to initial velocity data (non-linear regression fits enzyme kinetic data (initial velocity V0 vs. substrate concentration S to models like the Michaelis-Menten equation in Scheme 3a estimate Vmax and Km) to derive kcat and Km; followed by computing their ratio. This method necessitates error propagation and may lead to substantial standard errors (Scheme 4). Alternatively; you can rearrange the Michaelis–Menten equation to directly obtain kcat/Km; often resulting in lower error estimates. The decision to use kcat/Km; kcat or Km as comparative parameters hinges on the specific application. For biocatalytic applications; kcat is more fitting; whereas kcat/Km is favored in mechanistic studies. Investigating factors that influence kcat/Km can yield valuable insights into the initial stages of enzymatic reactions [18,42-45].
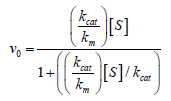
Scheme 4: A restructured version of the Michaelis–Menten equation allows us to calculate kcat/Km and kcat directly through non-linear regression analysis [32].
The Michaelis–Menten equation can be linearized to analyze enzyme kinetics effectively; with the Lineweaver–Burk plot (Figure 3) being the most common form; plotting the reciprocal of initial velocity against the reciprocal of substrate concentration (Scheme 3a). The slope indicates Km/Vmax; and the y-intercept reflects 1/Vmax. Other forms include the Eadie–Hofstee and Hanes–Woolf plots (Figure 4). Nonlinear regression software has minimized the need for linearization; providing reliable results with fewer data points and simplifying error assessment. However; linearized plots remain valuable for studying inhibition patterns and substrate interactions [12,35,37,46- 49].
Figure 3

Figure 4

[50] Enzyme catalytic properties can be affected by inhibitors; which can be competitive or allosteric. Competitive inhibitors bind to the enzyme’s active site; blocking substrate binding and reducing catalytic efficiency. In fully competitive inhibitions; both substrate and inhibitor are converted into products; while in partial competitive inhibition; the enzyme-inhibitor complex becomes inactive. Understanding the dynamics of these inhibitions is essential for developing drugs targeting specific enzymes. The efficiency of a drug depends on how variations in the inhibitor concentration affect the enzyme’s kinetic parameters; such as Km and Vmax. Kinetic parameters can be determined through steady-state experiments and non-linear least square fitting; though this may introduce high error levels in the resulting calculations [51]. The Dixon plot is a variant of the linearized Michaelis–Menten equation that integrates Kᵢ and I; enabling direct extraction of Kᵢ from a 1/v versus I plot at a constant substrate concentration. Substrate inhibition can be modeled with a modified Michaelis– Menten equation; and nonlinear regression is the preferred method for calculating Kᵢ; with many graphing software providing preset equations for this analysis [24,52]. Steady-state kinetic analysis reveals substrate binding and product release in multi-substrate enzymatic reactions. A double-reciprocal plot (1/v vs. 1/(S₁)) helps differentiate between sequential (random or ordered) and ping-pong mechanisms; parallel lines indicate a ping-pong mechanism; while intersecting lines suggest a sequential mechanism [30,38,40,53].
The steady-state kinetics approach offers advantages like higher signal amplitudes and fewer substrate data points but requires advanced data fitting compared to traditional Michaelis–Menten methods. It reduces uncertainties in kinetic parameter estimation through globally fitting progress curves; though fitting with integrated Michaelis– Menten forms is uncommon [38,30,54-56]. Fitting data; as demonstrated by Michaelis and Menten over a century ago; places considerable constraints on the quality of the data collected. This method limits data fitting to the initial phase of the curve where only minimal amounts of product are formed. A more precise approach involves monitoring the reaction over extended periods; allowing the reaction rate to decline as the substrate is consumed. This deviation from linearity can be addressed by utilizing computer simulations for data fitting. Additionally; product inhibition can be effectively managed if it plays a significant role in the data [57].
Transient-State Kinetics
The transient-state kinetics or pre-steady-state period is the brief phase that follows the initiation of an enzymatic reaction before steady-state conditions are reached [58,59]. Steady-state analysis simplifies enzymatic reactions by treating the conversion of the (ES) complex to product (E + P) as a single step; but it overlooks the individual steps at the active site. The transient-state period offers valuable insights into the enzyme’s mechanism; these methods enable the observation of specific events in a reaction pathway over short time frames. They allow monitoring of intermediate formations and conformational changes during a single turnover; thereby facilitating the determination of the kinetics of the elementary reaction steps [60- 62] (Figure 5). These steps are generally not visible in steady-state analysis.
Figure 5

Transient-state kinetics differ from steady-state kinetics by measuring rapid physical and chemical events at the active site within milliseconds rather than after the reaction has stabilized. This approach helps determine intrinsic rates and equilibrium constants for enzymatic reactions; including substrate binding; transformations; conformational changes; and product release. This detailed analysis of enzyme reactions reveals insights into reactive intermediates; transition states; conformational changes after substrate binding; and the rate-limiting steps in the catalytic pathway [32].
Steady-state kinetics can indicate the order of substrate and product binding and release but cannot accurately identify the specific elementary steps involved [61,62]. Steady-state kinetics are crucial for setting up transient-state experiments by determining concentrations and time scales. When combined with steady-state analysis; transient-state kinetics can pinpoint rate-limiting steps in chemical or physical processes. The value of kcat reflects the slowest step in the catalytic cycle after substrate binding; which is essential for enzyme engineering aimed at enhancing the specific rate-limiting step while minimizing changes to faster steps [13].Transient-state kinetics enables researchers to use substrate analogs; inhibitors; isotopically labeled substrates; or pH variations to gain direct insights into the active site’s physical and chemical changes and their rates [20,28,53,60]. Combining transient kinetics with steady-state analysis deepens our mechanistic understanding of enzyme catalysis by utilizing both kinetic and thermodynamic principles [61]. This approach allows comparison of how reaction conditions; modified substrates; and active-site mutations affect chemical and physical events in enzyme reactions [42,64-67].
[67] Transient-state experiments can generally be conducted using two primary approaches.
1. First Approach: This common method uses excess substrate compared to the enzyme; typically resulting in only a few turnovers during a brief observation period. Challenges may arise in separating overlapping spectroscopic signals; such as absorbance and fluorescence; when multiple turnovers occur simultaneously.
2. Second Approach: Single turnover kinetics use excess enzyme over substrate to observe only one turnover; enhancing the resolution of reaction steps without interference from later turnovers.
The ability to use either excess enzyme or substrate helps differentiate between saturation kinetics; which involve a non-covalent complex; and second-order kinetics in enzyme-substrate interactions. This distinction is especially valuable when studying oxygenase reactions with O₂ [68]. In experimental contexts; transient-state kinetics require rapid-mixing techniques to monitor reactions milliseconds after they start; often needing high enzyme concentrations for strong signals. The two main techniques are:
1. Stopped flow: Monitors reaction progress through changes in signals like absorbance or fluorescence.
2. Rapid-Quench-Flow: Quenches reactions after short intervals for analysis using methods like HPLC or LC-MS to quantify substrates and products [69].
Each method has unique benefits and can be chosen based on the properties of the products and intermediates analyzed. Lower temperatures slow reactions for better observation of intermediates and products. For more on stopped-flow and rapid-quench-flow experiments; see the following references [70-77]. Some effective but less common enzymology techniques; such as flash photolysis and relaxation methods (temperature; pH; or pressure jumps); avoid mixing hydrodynamics and enable monitoring in the picosecond and microsecond ranges [73,78]. Light-dependent enzymes are ideal for transient-state kinetic analyses with rapid-reaction techniques. They allow for mixing all reaction components before catalysis starts with a brief light pulse (flash photolysis); enabling monitoring of reactions on very fast timescales [79]. Transient-state kinetics may not directly apply to biocatalysis; but studying enzyme reaction pathways using rapid-reaction techniques is essential for understanding enzyme mechanisms. This knowledge aids in optimizing enzymes through protein engineering and highlights potential application challenges. Characterizing intermediate kinetics is crucial for isolating and analyzing them. Integrating structural information with mechanistic details is important for rational enzyme design and emulating native reactions. Without understanding enzymatic pathways; including transition states and intermediates; designing specific reaction enzymes is not feasible [80,81].
Analyzing transient-state kinetic experiment data requires integrating rate equations for each step. Fitting time-course data to analytical solutions is challenging due to complex rate equations; which often necessitate approximations; such as single- or double-exponential models [12,59,62]. Using numerical integration of rate equations from a proposed mechanism enhances kinetic resolution; yielding accurate estimates of rate and equilibrium constants; provided that sufficient data capture the mechanism’s complexity [59,62]. Accurate initial details on enzyme and substrate concentrations; response factors; and estimated rate constants are vital for effective least-squares fitting to a kinetic model. If time-course data from multiple experiments are available; this can be globally fitted to a unified mechanism to derive a consistent set of rate constants. Simulations can also predict rate constants; with the key difference that simulation parameters remain constant; whereas fitting allows them to vary. Various free and commercial software options exist for enzyme kinetic data fitting and simulations [62,80,81].
Data from steady-state kinetics can enhance analysis by considering the slowest step as kcat or using Km as an approximation of Kd [42,60]. Recent proposals include alternative numerical solutions for enzyme kinetic data analysis [53]. The use of transient kinetic methods to investigate enzyme mechanisms has surged due to recent advancements in instrumentation and the overexpression and purification of novel enzymes. Transient kinetics is increasingly favored for assessing site-directed enzyme mutants and for exploring intricate questions related to the connections between protein structure and observable function [82]. Coupled with advancements in structural and genetic analysis techniques; transient-state kinetic analysis lays the groundwork for what can be referred to as the “new enzymology.” However; a significant risk in interpreting kinetic data arises from the tendency to introduce an extra reaction step to explain an atypical kinetic outcome; when this new finding may be better explained by a simpler model that reflects a deeper understanding of kinetic subtleties [82].
The Rapid-Equilibrium Kinetics
Rapid-equilibrium rate equations for enzyme-catalyzed reactions are particularly valuable because; when experimental data align with these simpler rate equations; the Michaelis constants can be interpreted as equilibrium constants. However; for certain reactions; it becomes essential to utilize the more complex steady-state rate equations. In rapid-equilibrium derivations; thermodynamics is applied to determine the equilibrium concentrations of reactants leading up to the rate-determining step. This requires that the reactions within the mechanism be independent. This approach contrasts with steadystate mechanisms; where the independence of reactions is not a requirement [83].
Consider the reaction mechanism (Schame 1) involved in the enzyme- catalyzed transformation of substrate into product; with the understanding that the catalyzed rate significantly surpasses the non-catalyzed rate. Similar to the derivation of equations for facilitated transport reactions under rapid equilibrium conditions; this analysis relies on the assumption that the relative concentrations of the substrates; enzyme; and product can be determined by the dissociation constant Ks for the interactions; as well as the concentrations of each species during the initial phase of the reaction (i.e.; under initial rate conditions). Additionally; assume that the enzyme concentration remains constant over time [84]. The enzyme binds to the substrate with a second-order rate constant. The enzyme-substrate complex can dissociate with a first-order rate constant to revert to the substrate; or it can be transformed into the product with a first-order rate constant. If we assume that dissociation is much faster than the conversion of substrate to product; the relative ratios of substrate; enzyme; and product can be described accordingly [84]. Alternatively; consider this scenario: when the enzyme binds to the substrate; most of the complex dissociates; while only a small fraction is converted into product. If conversion occurs; the enzyme is freed and will quickly bind another substrate; reestablishing equilibrium; since the predominant fate of the bound substrate is dissociation rather than conversion to product. This reasoning aligns with the understanding that the physical step; characterized by dissociation; is likely faster than the chemical step involved in product formation [84].
Enzymatic Productivity
Enzymatic productivity quantifies product formation or substrate depletion over time; at a designated temperature and under specific reaction conditions. This metric reliably captures both the durability and reaction yield (conversion of substrate) of an enzymatic process; which are crucial for applications in translational research and biotechnology [85]. As a derivative measure; productivity is influenced by several factors; including:
• Enzyme activity and half-life
• Type and concentration of substrate
• Enzyme form and concentration
• Exposure time
According to [86]; enzyme kinetics are crucial for productivity under specific conditions. For instance; parameters like Km and kcat/Km are significant intrinsic factors; however; when (S) > Km; catalytic efficiency becomes less relevant. In cases involving artificial substrates with potentially high Km values; the specificity constant becomes the more pertinent variable. Moreover; neither kcat nor kcat/Km reflects the enzyme’s longevity on the time scales (hours/days) relevant to productivity analysis; as these metrics are derived from initial reaction rates measured over a few minutes. [86] Temperature influences enzyme activity in a multifaceted way. For example; the temperature optimum (Topt) is affected by the duration of the enzyme assay. Additionally; the melting temperature (Tm) reflects the structural unfolding (denaturation) of the enzyme and varies depending on the detection method used.
Far-UV circular dichroism monitors secondary structure; while intrinsic fluorescence; near-UV CD; and differential scanning calorimetry track the unfolding of tertiary structure. In all these techniques; the extent of stability (Tm) is influenced by the scan rate. Conversely; t1/2 assesses the activity regained after cooling and refolding the enzyme; which can impact productivity. Increasing temperature generally enhances enzyme activity up to a point; however; excessive temperature can reduce productivity due to enzyme inactivation [87]. Additionally; elevated temperatures have been associated with lower reaction rates for certain enzymes; stemming from irreversible unfolding caused by temperature-dependent transitions between active and inactive enzyme forms [88]. For some enzymes subjected to high temperatures; the active site may initially undergo local unfolding before the entire structure unfolds globally. Beyond temperature; enzymes can be destabilized by solvents; salts; pH; and other environmental factors. The activity–stability trade-off adds further complexity; suggesting that highly stable enzymes tend to be less active; while highly active enzymes—like cold-adapted ones—are more thermolabile [89,90]. Another critical factor affecting productivity is mass transfer limitations; which can affect the rate at which substrate reaches the enzyme’s active site. Mass transfer is influenced by several factors; including the type of substrate (simple vs. polymeric); the enzyme forms such as heterogeneous formulations (whole cells and immobilized); the viscosity of the reaction medium (which may change during the reaction due to product formation); the mixing efficiency of reaction components; and the method of substrate dosing [86].
Understanding how to optimize enzymatic activity is crucial not only for biotechnological applications but also for uncovering the fundamental principles that inform the design and enhancement of biological systems in nature. The Michaelis-Menten equation has served as a foundational framework for the study of enzymatic activity. However; clear guidelines for optimizing parameters to increase activity are still lacking [91]. Studies show that adjusting the Michaelis-Menten constant Km to match the substrate concentration (S) significantly increases enzymatic activity. This guideline; Km = (S); was derived mathematically on the premise that thermodynamically favorable reactions exhibit higher rate constants while the overall driving force remains constant. Bioinformatic analyses indicate that the relationship between Km and in vivo substrate concentrations is consistent across a dataset of nearly 1;000 enzymes; suggesting that even natural selection adheres to the Km = (S) [91]. Studies have been undertaken to translate in vitro and in vivo enzyme kinetics into pharmacokinetic and pharmacodynamic parameters. The goal is to identify drugs that either induce or inhibit enzymes; thereby informing optimal clinical efficacy and safety [92].
Because enzymes can influence the rates of biochemical reactions by selectively catalyzing specific substrates [93,94]. they are essential in metabolism; signal transduction; and cellular regulation; their dysfunction can lead to severe diseases [95,96]. Additionally; enzymes serve as highly specific catalysts in various industrial sectors; including drug development; biofuel production; and food processing [97]. For over a century; a foundational method for understanding enzyme kinetics has been the Michaelis-Menten equation; introduced by Michaelis and Menten [98] and later rigorously derived by Briggs and Haldane [99] using the standard quasi-steady-state approximation [100]. This equation illustrates how the rates of enzyme-catalyzed reactions depend on substrate concentration; utilizing two key parameters: the catalytic constant; kcat; and the Michaelis-Menten constant Km. The kcat signifies the maximum reaction rate at saturating substrate levels; Vmax= kcat ET; where ET represents the total enzyme concentration; while Km denotes the substrate concentration at which the reaction rate is half of Vmax.
To estimate kcat and Km from the measured product accumulation over time (i.e.; the progress curve); two primary assays are utilized: the initial velocity assay (initial rate analysis) and the reaction progress curve assay (progress curve analysis) [101-105]. In the initial velocity assay; reaction rates are recorded across a range of substrate concentrations. Subsequently; a linear transformation of this data; such as Lineweaver-Burk plots; enables straightforward estimation of the two parameters without computational tools [101,102]. Recent advancements in computational tools have introduced an alternative method: the reaction progress curve assay. In this approach; the complete time course (i.e.; progress curve) is fitted to the solution of a differential equation or an integrated rate equation; thereby making more efficient use of the data than the initial velocity assay [103,104,106]. Although more technically demanding; the progress curve assay requires less data to estimate parameters than the initial velocity assay.
Since both assays rely on the MM equation; they should be conducted only when the MM equation holds; that is; when the enzyme concentration is significantly lower than the sum of the substrate concentration and Km [100,107] Since the Km value is typically unknown beforehand; in vitro experiments usually utilize an enzyme concentration that is much lower than the substrate concentration to maintain the validity of the MM equation [108]. However; such conditions cannot be reproduced in vivo; as endogenous enzyme levels are typically much higher than those used in standard in vitro assays [109,110]. Consequently; applying the MM equation to analyze vivo data and predict enzyme activity based on parameters obtained from in vitro assays can be risky [108,111]. Moreover; even when the MM equation is applicable; precise estimation is not guaranteed due to the highly correlated structure and unidentifiability of the parameters [112-116]. Although estimated parameters may fit the data well; they can differ significantly from the actual values of kcat and Km. Because of the identifiability challenge; experimental designs aimed at extracting the maximum possible information about the parameters have been explored [105,106,113-117]. For example; to ensure that parameters can be identified from the initial velocity assay; the initial substrate concentration should be increased from a low level to a higher level until the reaction velocity reaches saturation. Generally; for saturation; the initial substrate concentration should exceed 10 Km; however; achieving such high concentrations can often be problematic [117]. For the progress curve assay; it is recommended that the initial substrate concentration be similar to Km [116,118]. It is important to note that both assays require prior knowledge of Km; leading to the paradox that; to estimate Km; an approximate value must already be known.
Enzyme Targeting in Drug Development
Enzyme kinetics help researchers understand complex biochemical pathways in diseases; identifying key points for therapy. This knowledge aids in designing drugs that effectively target and modulate enzyme activity for various ailments. Enzymes demonstrate substrate specificity through their ability to bind to molecules. Understanding enzyme kinetics is crucial in recognizing these specificities; which plays a vital role in drug development aimed at targeting disease-related enzymes. Additionally; examining enzyme inhibition kinetics contributes to the design of targeted medications that can effectively modulate enzyme activity [119].
Enzyme kinetics is crucial in drug discovery; facilitating breakthroughs in therapeutic innovation. Understanding enzymatic reactions allows for the precise design of drugs [120]. Understanding the interactions between enzymes and substrates; researchers can design targeted drugs for specific biological pathways [121]. This understanding is crucial in drug discovery; enabling scientists to design medications that interact precisely with specific enzymes or proteins [122]. Enzyme kinetics accelerates this process by providing insights into the pharmacokinetics and pharmacodynamics of drug candidates; optimizing enzyme-substrate interactions; and thus shortening the time from research to clinical use [123]. Enzymes involved in disease progression are key targets for many medications; which aim to inhibit them. Drug design begins with extensive screening of diverse chemical libraries to identify new inhibitors and bioactive compounds. Pharmaceutical companies perform over 50 million biological assays annually; including enzyme assays. However; creating reliable enzyme assays for identifying lead compounds remains a significant challenge in early drug development phases [119-123].
Inhibition of Neurotransmitter Degradation in Neurodegenerative Disorders: The breakdown of neurotransmitters is crucial for brain function; particularly in signaling between neurons. Acetylcholinesterase (AChE) efficiently terminates neuromuscular signaling by hydrolyzing acetylcholine; which is vital for cognition. In Alzheimer’s disease (AD); cholinergic signaling declines; leading to the use of acetylcholinesterase inhibitors (AChEIs) as the primary treatment for cognitive deficits. Butyrylcholinesterase (BChE) may also be targeted for cognitive decline. Additionally; the loss of other neurons in AD affects dopamine and serotonin levels; with MAO inhibitors used as antidepressants and for delaying Parkinson’s disease. ChEs and MAOs are key targets for developing multi-target drugs in neurodegenerative disease research [123-131].
Alpha-Glucosidase Inhibitors Promising Candidates for Type 2 Diabetes Treatment: Alpha-glucosidase inhibitors (AGIs) help lower blood glucose levels; potentially preventing or delaying type 2 diabetes mellitus (T2DM) and its complications in at-risk individuals. Currently; three AGIs used in clinical practice are acarbose; miglitol; and voglibose (Figure 6) [132]. The gastrointestinal tract’s degradation reactions convert complex carbohydrates into monosaccharides for absorption in the small intestine. This process starts with amylases from the pancreas and salivary glands; which hydrolyze starch into shorter polysaccharides. Pancreatic amylases further break down these polysaccharides in the small intestine. The final digestion phase involves α-glucosidase (AGI) in the enterocyte brush border; which hydrolyzes disaccharides and oligosaccharides. AGI is overexpressed in non-insulin-dependent diabetics; leading to higher post-meal glucose levels. Inhibiting α-amylase and AGI can delay glucose release; aiding type 2 diabetes management. Plant-derived α-amylase inhibitors; particularly from beans; show promise by mimicking carbohydrates to bind to the α-glucosidase enzyme. Various detection methods; including colorimetric assays; verify α-glucosidase inhibition by measuring p-nitrophenol release [133-137].
Figure 6

The prevalence of diabetes; particularly type 2; has increased due to aging; urbanization; and unhealthy lifestyles; resulting in obesity and decreased physical activity. Type 1 diabetes is characterized by T cell-mediated destruction of insulin-producing pancreatic beta cells; while type 2 diabetes stems from the body’s inability to manage sugar effectively; leading to high blood sugar levels and various complications. Type 2 diabetes; previously known as adult-onset; is now increasingly seen in children due to rising obesity rates. Although there is no cure; management through weight loss; diet; and exercise is essential. If lifestyle changes fail; medications like alpha-glucosidase inhibitors; metformin; and SGLT-2 inhibitors may be prescribed [138].
Enzymes play a crucial role in biotechnology by increasing reaction rates through lowering activation energy without being consumed in the process. Their effectiveness under mild conditions makes them invaluable across diverse industries; including biofuels and pharmaceuticals. Acting as essential biological catalysts in metabolic processes; enzymes underscore the importance of examining their kinetic properties for applications in enzymology; medicine; and synthetic biology. Analyzing catalytic rates and binding affinities is vital for drug development and improving biocatalytic processes. Environmental conditions; such as pH and temperature; significantly influence enzyme activity; each enzyme has an optimal pH and temperature at which it functions best. Deviations from these optimal conditions can affect enzyme activity; with extreme conditions leading to irreversible denaturation.
The Michaelis–Menten equation serves as a foundation for enzyme kinetics; enabling research into complex reactions such as multi-substrate and inhibition kinetics. Most enzymes conform to this model; which describes the reversible formation of an enzyme–substrate complex (ES) and its conversion to products at a specific rate. Steady-state kinetics involves measuring initial reaction rates at stable enzyme levels while varying substrate concentrations; which are generally much higher than the enzyme concentration. This leads to a state in which the formation and breakdown rates of the (ES) complex are nearly equal; allowing pseudo-first-order kinetics during initial velocity measurements. The transient-state kinetics; or pre-steadystate period; occurs right after an enzymatic reaction starts and before steady-state conditions are reached. While steady-state analysis simplifies reactions by treating the conversion of the (ES) complex to products as a single step; it overlooks the detailed individual steps of the active site. The transient-state period offers insights into the enzyme’s mechanism; enabling observation of reaction pathway events; intermediate formations; and conformational changes during a single turnover—details often missed in steady-state analysis.
Rapid-equilibrium rate equations for enzyme-catalyzed reactions facilitate interpreting Michaelis constants as equilibrium constants when experimental data fit these simpler equations. Some reactions; however; necessitate more complex steady-state rate equations. In rapid-equilibrium thermodynamics; equilibrium concentrations of reactants are established before the rate-determining step; assuming independent reactions; this requirement does not hold in steadystate mechanisms. Finally; enzymatic productivity gauges product formation or substrate depletion over time under controlled conditions; reflecting both durability and reaction yield. This is essential for translational research and biotechnology. Optimizing enzymatic activity is critical for biotechnological applications and for enhancing our understanding of biological systems; with the Michaelis-Menten equation.
In conclusion; enzymes are pivotal to advancing biotechnology due to their ability to accelerate reaction rates. Their utility across various industries; such as biofuels and pharmaceuticals; is primarily due to their efficacy under mild conditions. Understanding enzyme kinetics through models such as the Michaelis–Menten equation is fundamental for exploring complex biochemical reactions and optimizing enzymatic processes. Analyzing both steady-state and transient- state kinetics provides a comprehensive understanding of enzyme mechanisms; enabling enhanced drug development and biocatalytic improvements. Furthermore; recognizing the environmental factors that influence enzyme activity is essential for maintaining optimal function and preventing denaturation. Overall; optimizing enzyme activity is crucial for both practical biotechnological applications and deepening our comprehension of biological systems; positioning enzymes as central to current and future scientific advancements.

























