info@biomedres.us
+1 (502) 904-2126
One Westbrook Corporate Center, Suite 300, Westchester, IL 60154, USA
Site Map
Received: August 07, 2024; Published: September 16, 2024
*Corresponding author: Orchidea Maria Lecian, Sapienza University of Rome, Rome, Italy
DOI: 10.26717/BJSTR.2024.58.009189
Some of the Kirsten rat sarcoma viral oncogene homologue (KRAS) are newly analysed. The qualities of the KRASG12D proteins are investigated. In the case of interaction with generic membranes, the corresponding new Hidden Markov-State model is newly defined. The Kolomogorov backward equation and the Kolomogorov forward equations are thereafter considered. The clustering method of the states of the Hiddedn Markov-States Model (HMsM) is one based on the analysis of the root mean-square deviations; the fundamental matrix of the chain is defined. The relevance of the study of the standard deviation in the choice of the representation of the probability matrix is newly studied.
Keywords: Krasg12d; Markov Fundamental Matrix; Markov Probability Matrix; Hidden Markov-States Models; Non-Markov Processes
Abbreviations: HMsM: Hiddedn Markov-States Model; KRAS: Kirsten Rat Sarcoma Viral Oncogene Homologue; HVR: Hyperviable Region; MM: Markov Model; Ras: Rat sarcoma Virus; GDP: Guanosine Di-Phosphate; GTP: Guanosine-5’-Tri-Phosphate
Kirsten rat sarcoma viral oncogene homologue (KRAS) proteins are understood as [1] ’drivers’ in human cancer. The quantification of their qualities have rendered has rendered possible to infer the biological timescales of the processes sampled. The studies can be conducted also at atomic level. The different KRAS proteins are discriminated according to the properties of their hyperviable regions (HVR). The role of KRAS proteins is understood as signal-transducer proteins of the Hyper-Viable Regions (HVR’s) of the pathways. The phenomenon can happen both either as a direct Ras mutation, or according to the proteins in the Ras pathways. The speed of the diffusion of the Ras proteins in membranes are used for signaling the response time in the activation of Raf and in that of PhosphatidylInositol 3-Kinase (PI3K) [2]. The speed of the response time in the activation is relevant for understanding whether all the pathways are ’affected’ in all the coordinates.
In the present paper, the interaction patterns of the KRASG12D proteins are recapitulated; the behaviours are outlined, which allow one to define a new Hidden Markov-State Model in which the clustering methods are based on the analysis of the variances. The oroginating (Markov) chain is newly inspected. The questions about the choice of the representation of the probability matrix for the Markov Model (MM) is here discussed within the obeyance of the Kolomogorov backward equations and to that of the Kolmogorov forward equations according to the experimental data accounting for the variances. The study can be apt for a crisp landscape of the MM, where the states are defined on opportune Borel subset. The analysis is pertinent with the confrontation with possible non-Markov behaviours The present study is relevant within the most recent understanding of new properties of the KRAS12D proteins, as pointed out in [3].
The understanding is proven, that the KRAS interact with the
lipidic membranes allosterically [4]; indeed, the binding of the Rat
sarcoma virus (Ras) proteins demonstrates a perturbation effect
on the conformational ensembles of its ’effectors’ [5]. Inhibitors of
KRAsG12G have begun being studied [6]; for this purpose, allatoms
molecular dynamics has been attempted to be codified within MarkovState
Models (MSM’s): more in detail, stationary probabilities can
be derived for the selected MSM. The AMG 510 is an effective selective
inhibitor which is apt for interaction with non-small cancer cells. The
design of KRAS inhibitors can therefore be pursued; for these purposes
the covalent bonds between the compound of the thiol groups
from the cystine residue are studied [7]. MSM’s are used to analyse
the trajectories from the molecular dynamics. Guanosine di-phosphate
(GDP) bounds and Guanosine-5’-tri-phosphate (GTP) bonds of
KRAS are studied. The effective MSM is one obtained after clustering:
the clustering method apt for describing the dynamics is one which
selects the states according to the rootmean-square deviation. The
MSM is constructed according to the gathering of the several metastable
states after the use of ’defined collective variables’. A transition
probability matrix 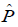 is this way constructed, which obeys the
forward equation.12 clusters are issued which define the states Xi, i
= 1,...,12, which gather the trajectories: probabilities of transition between
states are found from the entries of
.
is this way constructed, which obeys the
forward equation.12 clusters are issued which define the states Xi, i
= 1,...,12, which gather the trajectories: probabilities of transition between
states are found from the entries of
.
The stationary populations of the states are calculated. It is one of the purposes of the present study to develop the tools to calculate the variation in time of the population(s) of the states; the compatibility with the biological timescales will allow for the comparison with possible properties of non-Markovianity. The differences between the properties of GTP and those of GDP are outlines in the differences between populations of selected states. From [8] and form [9], the difference between ’active’ conformations and ’inactive’ conformations are found in the interaction between the γ-phosphate of the GTP form the Switch 1 region and that of the Switch 2 region. In active conformations, the Switch 1 and the Switch 2 are ’stabilised’. For inactive conformations, the loss of interaction of the γ-phosphate is described. The drug MRTX1133 is known as potential inhibitor of KRASG12D. The structural binding paradigms of MRTX1133 are studied in [10]. MRTX1133 stabilises the sites of the binding after the increase of hydrophobicity, which modifies the movements of Switch 1 and those of Switch 2.
From the results from [6], the states contained in each cluster are
selected according to the root mean-square deviation; in the analysis
of [4], clusters of states are selected according to the k-mean method
[11], according to which a Hidden Markov State Model (HiddMSM)
is built. The Kolmogorov backward equation must be imposed for a
Markov Model to hold. The fundamental matrix ![]() needs to be defined; it is found from the request that the possible transition matrices
needs to be defined; it is found from the request that the possible transition matrices
![]() of the possible chains obey the backward equation
of the possible chains obey the backward equation
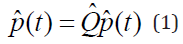
where the p(t) transition matrix is one of the possible transition matrices.
In the present case, the analysis of the variances allows one to
define the HiddMSM with a ![]() . The main interest in the new HiddMSM
is the relevance of the roles of the states X11 and X12; in addition to the
calculation of the stationary populations, the passage times from the
state X11 to the state X12 can be calculated from the eigenvalues of
. The main interest in the new HiddMSM
is the relevance of the roles of the states X11 and X12; in addition to the
calculation of the stationary populations, the passage times from the
state X11 to the state X12 can be calculated from the eigenvalues of ![]() (i.e. as from [12]). The new HiddMSM is therefore one consisting of
the final state where the state X11 and the state X12 are clustered; the
resulting state from the Markov landscape is one Borel subset. The
construction of this HiddMSM is allowed according to the possibility
to build one single state after the clustering of the two previous-mentioned
states; this possibility is validated after the control of the
variances. The choice of the representation of the probability matrix
importantly depends on the entries of the fundamental matrix. The
obeyance of the Kolmogorov backward equation is controlled accordingly.
The eigenvalues can therefore be calculated. The eigenvalues
are once more defined after the control of the variances of the two
clustered states.
(i.e. as from [12]). The new HiddMSM is therefore one consisting of
the final state where the state X11 and the state X12 are clustered; the
resulting state from the Markov landscape is one Borel subset. The
construction of this HiddMSM is allowed according to the possibility
to build one single state after the clustering of the two previous-mentioned
states; this possibility is validated after the control of the
variances. The choice of the representation of the probability matrix
importantly depends on the entries of the fundamental matrix. The
obeyance of the Kolmogorov backward equation is controlled accordingly.
The eigenvalues can therefore be calculated. The eigenvalues
are once more defined after the control of the variances of the two
clustered states.
The behaviour of the KRASG12D are newly analysed with the purpose
of isolating the properties which allow one to construct Hidden
Markov-States models, whose state are issued according to the analysis
of the variances. The forward equation is obeyed by the probability
matrix P; the request that the backward equation be obeyed requires
the definition of a fundamental matrix ![]() of the chain, that, on
its turn, allows one to define a host of possible transition matrices
of the chain, that, on
its turn, allows one to define a host of possible transition matrices ![]() , from which the transition matrix apt for the model is scrutinised according
to the analysis of the variances. The calculation of the eigenvalues
allow one to estimate the Mean Fisrt Passage Times (MFPT’s)
and their time evolution. The further perspectives are the study of the
changes in population in times of the states Xi within the biological
timescales for the dynamics to be validated; the possible chains have
to be discussed within the (propagation of the) statistical errors. The
compatibility of different timescales within the Markovian features of
the model is still an open problem.
The new HiddMSM is therefore to be analysed as far as the time
evolution of the eigenvalues is concerned. The procedure allows one
to reconstruct the time evolution of the errors according to the Markov
modellisation chosen, and, in case, on the discretisation scheme
adopted, i.e. such as (but not necessarily only) the coarse-graining
methods. It is therefore possible to trace back the pathways of the
Markov landscape(s) (i.e. the paths on the chosen Borel subset).
, from which the transition matrix apt for the model is scrutinised according
to the analysis of the variances. The calculation of the eigenvalues
allow one to estimate the Mean Fisrt Passage Times (MFPT’s)
and their time evolution. The further perspectives are the study of the
changes in population in times of the states Xi within the biological
timescales for the dynamics to be validated; the possible chains have
to be discussed within the (propagation of the) statistical errors. The
compatibility of different timescales within the Markovian features of
the model is still an open problem.
The new HiddMSM is therefore to be analysed as far as the time
evolution of the eigenvalues is concerned. The procedure allows one
to reconstruct the time evolution of the errors according to the Markov
modellisation chosen, and, in case, on the discretisation scheme
adopted, i.e. such as (but not necessarily only) the coarse-graining
methods. It is therefore possible to trace back the pathways of the
Markov landscape(s) (i.e. the paths on the chosen Borel subset).

























