info@biomedres.us
+1 (502) 904-2126
One Westbrook Corporate Center, Suite 300, Westchester, IL 60154, USA
Site Map
Received: February 03, 2023; Published: February 24, 2023
*Corresponding author: Guojun Zheng, State key laboratories of chemical Resources Engineering Beijing University of chemical technology, Beijing 100029, China
DOI: 10.26717/BJSTR.2023.48.007721
The highly transmissible COVID-19, originated at Wuhan city China in December 2019 has rapidly spread with millions of confirm cases. Identification of potential drug is an urgent need to combat this pandemic is an urgent global need. To date, it has affected 647,972,911 individuals in more than 200 countries and has been responsible for 6,642,832. Insilico therapeutics are urgently needed to cure patient severely infected with Covid-19. Drug repurposing is usually applied in Insilco drug design has a number of benefits due to its time and cost-efficient strategy, which fundamentally entails the identification of new targets for existing therapeutic candidates. Here, we performed a computational drug designing strategy to investigate candidates for RdRp protein of CoVID-19 inhibitors, searching for druggable cavity pockets within the viral protein. Using molecular docking from a commercially available Chembridge database, we performed pharmacophore-based virtual screening to identify potential hits against RdRp. The Lipinski rule of five was used to identify 845 hits from the Chembridge database through pharmacophore modeling. The top 283 compounds were chosen after these hits were docked into the binding site of (RdRp) NSP12 and evaluated for docking scores. 10 compounds were further chosen from the database based on the best ligand interactions. Finally, three lead compounds were chosen using a variety of computational analysis techniques such as the MMPB(GB)SA analysis, and root mean square deviation and fluctuation. According to the Lipinski rule of five, all of the compounds demonstrated good ADMET properties, indicating that their roles had optimal properties. The three compounds that were found may be useful against CoVID-19, but more research is required to provide conclusive proof. We think that the findings of this research may present a novel method for the development of COVID-19 avenue.
Keywords: SARS-CoV-2; Homology Modelling; Virtual Screening; Homologous Structure; Silico Pharmacophore; Drug; Hydrogen Bond; Energy; Molecular
Abbreviations: PSI: Position Specific Iterated; PHI: Pattern Heat Initiated; RMSD: Root Mean Square Deviation; DON: Hydrogen Bond Donor
The COVID-19 pandemic has led challenging circumstances all around the world. Epidemiology, testing, clinical care, prevention, management, and the rate of therapeutic drug discovery have all advanced at a faster pace [1-5]. Drug discovery is a time-consuming and expensive process that involves long evaluation of clinical trials, has a minor success rate, and averages between $2-3 billion [6,7] and also fails to deliver on time [8]. Drug repurposing, as opposed to drug invention, has a number of benefits, one of which is that it can be administered during pandemics. The global pandemic SARS-CoV-2 wreaked havoc on the global economy and public health [9,10]. The risk of suffering more severe COVID-19 problems appears to be higher in older adults and those with significant underlying medical illnesses, such as diabetes, heart disease, or lung disease. The new SARS-CoV-2 has spread widely since it first appeared in Wuhan, China, in December 2019 [11]. At present, there are 647,972,911 confirmed cases of COVID-19 and 6,642,832 deaths reported to the WHO. The total number of newly reported cases in the last 14 days is 3,347,311; the number of newly recorded deaths is 9,507; and the number of infections is doubling every 2-10 days (https://covid19.who.int/table). Due to the sudden and large rise in COVID-19 cases, the WHO is calling for "immediate, strong efforts" to fight the viral pandemic [12]. The WHO designated the COVID-19 outbreaks as the "sixth public health emergency of worldwide concern" on January 30, 2020 [13].
The significant increase in COVID-19 infections urgently calls for the rapid development of medications. A quicker and less expensive method for repositioning medicines against new targets is computer-aided drug design [14]. The strategy includes exploring novel targets for the known therapeutic options. Positive-sense, single-stranded RNA coronaviruses have a 30 kb genome, a 50 cap structure, and a 30 polyadenylated tail [15]. The ORF1a and ORF1b polyproteins that their genomes code for carry out a variety of tasks, including attaching to and penetrating the host cell, viral replication, and evading the host immune system. The coronavirus proteins aggregate the replication-transcription complex and a number of other structural proteins into a heteromeric complex to carry out their intended function [16,17]. One of these subunits is the non-structural protein 12 (NSP-12), which involves RNA-dependent RNA polymerase (RdRp) activity. Numerous studies have focused on the RDRP protein as a potential antiviral target to prevent the spread of multiple viral illnesses, such as dengue, hepatitis C, zika, West Nile disease, and Japanese encephalitis [18,19]. In this study, we used an in silico drug repositioning technique with the ChemBridge database to find compounds that inhibit the RNA-dependent RNA polymerase (RDRP) of CoVID-19. The RDRP protein has been looked at as a possible way to treat many illnesses caused by viruses. This entails using an existing medication to treat a condition other than the one it was developed to treat.
As the pharmacokinetics of these medications are already established, phase I clinical trials are not required, which cuts down on the time and expense of drug discovery which is absolutely necessary in the current global environment. Despite the urgent need for the quick identification of inhibitors against CoVID-19, it is crucial to guarantee the security of the suggested medications or vaccinations. Drug repositioning is a safer way to find inhibitors against SARS-CoV-2 because scientists already know how medicines work and if they are safe. Currently, many studies including single-ligand docking [20,21] as well as screening of the compounds library [22] have been conducted to search for potential inhibitors against NSP-12 of SARS-CoV2. In our study, we employed the structural-based homology modeling method to identify structurally conserved proteins and then identified FDA-approved inhibitors of the homologous structure. Dasabuvir sodium [23,24] was chosen as an FDA-approved inhibitor because it had previously been approved for inhibiting HCV NS5B [23,24]. Furthermore, the conserved active site of NSP-12 was characterized, and the affinity of the inhibitors was evaluated using in silico pharmacophore-based virtual screening, molecular docking, and simulations.
Homology Modeling of RDRP Protein of SARS-CoV–2
The x-ray crystal structure of RDRP protein of SARS-CoV–2 was not available, so its homology model was constructed by an online server I-TASSER for further study. The sequence of the protein (NCBI ID YP_009725307.1) was obtained from the online NCBI server in FASTA format. The sequence was then uploaded to I-TASSER server [25]. It gives five homology models; these five models were checked by different validation tools.
Homology Model Validation
ERRAT server [26], Verify3D servers [27], Rampage and Ramachandran plot were used to check the overall quality of the model. By using rampage (MOE package) the stereo chemical quality of the model was assessed. Based on good scores from these tools one best model was selected for further study.
Exploration of Drug Like Features
A pharmacophore based on Ribavirin was designed for exploring drug like features in the compound leading to the inspection of large chemical libraries in MOE 2020. Ribavirin was first rigidly docked into the active site of Ribavirin followed by pharmacophore generation with Extended Hueckel Theory (EHT) approach. Pharmacophore was generated with seven features in which three were marked as essential features. The structure of the NSP12 of SARS-coronavirus (PDB ID: 6NUR) was selected as the template for modelling the NSP12 of SARS-CoV–2. The generated model was validated by a test database containing SARS-CoV–2 along with Ribavirin. All compounds of the test database were screened on the seven-featured ligand-based pharmacophore and their mapping modes were analyzed. For evaluation and assessment of drug like features, the validated pharmacophore geometry was screened with chemical libraries like ChemBridge database using screening protocol implemented in MOE 2020. Such model is used as 3D query in in-silico screening to identify hits of different physic-chemical properties The main purpose of such screening is to identify a novel drug like pose for further assessment. 283 hits were retrieved from ChemBridge database as a result of screening. To predict the drug-likeliness of compounds, Lipinski rule of 5 were applied, according to which the compound to be drug like would have log P in range of −0.4 to +5.6, molecular weight 180 to 500 Daltons, number of atoms from 20 to 70 (includes H-bond donors not more than 5 and H-bond acceptors not more than 10. PSSolar surface area no greater than 140Å [28]. Strictly following the above rules finally, 3 hits of Chembridge database were selected for further evaluation.
Molecular Interaction Study and Selection of Leads Compounds
For molecular interaction studies and selection of lead compounds, all the retrieved hits were docked into the binding site of RDRP (NSP12) protein of SARS-CoV-2. Docking protocol implemented in MOE 2016 with parameters like rigid and ligand based docking was performed. A maximum of 5 conformations were allowed to be saved for each ligand using the default parameters of MOE i.e. Placement: Triangle Matcher, Rescoring: London dG, GBVI/WSA dG, Refinement: Rigid Receptor; Using SVL script of MOE the root mean square deviation (RMSD) between the co-crystallized and re-docked conformation was calculated which was equal to 0.78 Å suggesting that our docking protocol is reliable [29]. On the basis of docking score, 3 top ranked compounds were selected for further evaluation. All the 3 compounds having better or at least comparable binding affinity and binding energy to the reference compound were selected for molecular dynamic simulations. The docking score, binding mode, pharmacophore mapping, binding energy (stability), binding affinity, and visual ligand interaction indicate that these selected lead compounds might act as structurally diverse, potent and novel antagonist for RDRP (NSP12) protein of SARS-CoV-2.
Binding Cavity Measurement
To understand the effect of the mutation on the ligand binding, cavity analysis was carried out. The online server, CASTp, was used to determine the changes in the volume of the binding cavity of native as well as mutants SMT. Alpha shape method is used by this server to measure volume and area of cavities. This server clearly distinguishes the inner and outer surface pockets. The default value, 1.4Å, for probe radius was used. The detected pockets were ranked based on the area and volume.
Molecular Dynamic Simulation of RDRP
The protein physiological state information is limited in the rigid body docking of X-ray crystal structure in static condition. Further to modulate the desired targeted protein function and detailed information is required. Molecular dynamics software can pave the way non-ideally to the detailed information. The biological complex dynamics solvent condition can be provided with these tools and the probable movements of a domain and its conformational changes can be predicted. All the simulations were carried out on a Linux (openSUSE) GPU based, workstation, using an AMBER22 software package [30]. All eight protein systems were simulated for 100ns. To maintain system neutrality, counter ions (Cl¯ and Na+) were added. Hydrogen atoms were added to the crystal structures of both enzymes, by the LEAP module. Truncated octahedral box of OPC water model with 10 Å buffer were used for solvation of all the protein systems. To study long-range electrostatic contacts, PME method (Particle Mesh Ewald) [31] was used. For all simulations, the ff19SB force field was used [32,33]. All the bonds including hydrogen atoms were constrained with the SHAKE algorithm [34]. For acceleration of MD simulations, the CUDA version of PMEMD was used [35]. Two steps energy minimization protocol was used for each solvated system minimization: the conjugate gradient minimization step for 20000 cycles and the steepest descent minimization step for 10000 cycles. The minimized systems were then heated up to 300K for 400ps.
Consequently, at a constant pressure of 1atm and temperature of 300K, each system was equilibrated. At the first step, the density was equilibrated with weak restraint for 50ps. Next, the system was equilibrated without any restrain for 2000ps in the NVT ensemble. Lastly, the production step was run under the NPT ensemble at 300K for the 100ns. To control the temperature, the Langevin thermostat was used [36].The coordinates for each system were stored at an interval of 2ns for consequent analysis. The RMSF, RMSDs, and other analyses like MMGBSA and hydrogen bond analysis between ligand and receptor were carried out. The analysis has been pursued by the CPPTRAJ module in the Amber20 program and AmberTools19 using trajectory files of each simulated system. The Origin Pro software [37] and GnuPlot [38] were used for the graphical visualization and interpretation of output files from the CPPTRAJ module [39], where MOE2016 [40] and Pymol v2.0 [41] software was used for protein visualization.
Hydrogen Bond Analysis
Using Amber20's CPPTRAJ package, we conducted an H-bond analysis to gather more details [42]. In order to comprehend the variations brought on by the substation of residues RDRP (NSP12) protein of SARS-CoV-2, the total number of H-bonds is determined. 10000 frames were taken during the MD simulation to assess the diversity among the ligand-protein complexes of the Reference drug, and the identified hits was employed to examine the hydrogen bonds between the protein-ligand targets. H-bonding is described in this paper as occurring at a distance of 3.5. All results are computed using the original program.
Binding Free Energy Calculations (MM/GBSA)
To predict quantitatively protein ligand binding energies, MM/GBSA method was used. This method is frequently used to accomplish classical MD simulations combined with free energy calculations [43]. 500 snapshots were sampled from the last 5ns trajectory, for the estimation of binding free energy. The MMPBSA.py script implemented in the Amber program was utilized for binding free energy calculations of all simulated SMT proteins (eight proteins).
According to the following equations, based on MM/GBSA method binding free energies (<ΔGbind) are calculated:
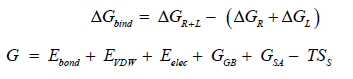
In the above 1st equation,
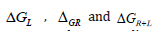
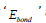
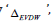

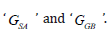
Homology Modeling
The experimental 3D structure of RDRP protein of SARS-CoV–2 is not available. Therefore, Homology modeling were carried out to elucidate of the 3D structure I-TASSER. The tertiary structure of RDRP protein was modelled with homology modelling tool, resulting top 10 independently identified threading templates was generated having the best Z score, which indicated a good alignment. The predicted 3D structure was thus expected to be highly accurate [45]. The homology model was created using the 3D structure of the human RDRP protein from the SARS-CoV-2 proteins (PDB code 6NUR), which exhibits the highest level of sequence identity with the query protein sequence at 96%. Utilizing a variety of structural evaluation techniques, the validity of the model was examined, including the geometric characteristics of the backbone conformations. The model was validated by RAMPAGE and ERRAT to check the quality of the modelled structure. Then the ProSA-Web services was used to confirm the protein structural accuracy with the predicting the Z-score of -11.44, the typical range for proteins of the same size found in nature [46,47]. The position specific iterated (psi) against pattern heat initiated (phi) distribution values of non-glycine and non-proline residues on Ramachandran plot are shown in (Figure 1 & Table 1). The Ramachandran plot showed that 79.9% in favored region, 17.4% of residues in allowed regions, and 0.9% residues were found in the disallowed regions. The red area in the graph indicates the most allowed regions while yellow region shows the allowed regions. ERRAT showed overall quality factor 95%. Both of these results indicate that the generated RDRP protein of SARS-CoV–2 model is valid with good stereo chemical quality. To find out the conformational variations, the template structures and RDRP protein of SARS-CoV–2 model were superposed and it was found that it indicated the close structural identity with their RMSD values 0.345Ao (Figures 2a & 2b). The validated model used further in this study.
Figure 1 A. Model of the RDRP (NSP12) protein of SARS-CoV-2 that was constructed by homology modelling. B. Superposition of homology model (magentas) and template SARS-CoV nsp12 (yellow) proteins.

Table 1: Ramachandra plot statistics of RDRP NSP12 protein.

Figure 2 Validation and refinement of the final vaccine using web resources A. Structure validation with a Z-value of -11.44; R. plot of the predicted model.

Development and Validation of Structure-Based Pharmacophore Model
The pharmacophore model describes an arrangement of chemical functional groups in space that are present in different compounds and are noticeably involved in interactions with the active residues of the active-site and therefore also required for biological functions. In ligand-based pharmacophore modeling, it is predictable that when compounds have the similar structure, will also have the similar biological activity as well as the interactions/binding with the drug target protein [48]. A common pharmacophore model based on Ribavirin was generated using MOE. A seven features pharmacophore model having two hydrogen bond acceptors (Acc), one hydrogen bond donor (Don), one hydrophobic (Hyd/HydA), one aromatic atom (Aro), and two Don&Acc was developed using default parameters (Figure 3). The developed pharmacophore model was validated by using a test database having 816 known inhibitors having 16 active and 800 generated decoys (inactive/least active) [49]. The test database was screened on the developed pharmacophore model. Interestingly, all the 16 active compounds were predicted as active compounds whereas 800 inactive compounds were shown to be inactive by the developed pharmacophore model. The obtained results reflect the accuracy of our generated pharmacophore model (Figure 3).
Figure 3 Three-dimensional pharmacophores feature generated from complex structure of proteins and Ribavirin by MOE. (b) Overlay of Ribavirin (triple inhibitor) on the pharmacophores features.

Database Screening
Finally, commercially available database, ChemBridge compound libraries were screened based on the generated pharmacophore. Consequently, we have found numerous compounds having stronger interactions with the target. Assessment of Drug-Like Features with Chemical Libraries. Subsequently, 845 structurally diversed hits were retrieved as a result of screening from ChemBridge database. To predict the drug ability of initial hits, these hits were filtered on Lipinski’s rule of five. This rule describes that drug like compounds should have logP value less than 5, molecular weight less than 500 Da, hydrogen bond donors less than 5 and hydrogen bond acceptors less than 10, otherwise they have poor absorption or permeation [50]. As a results of this filtering, 283 hits were predicted as drug like molecules. These molecules were docked in the active sites of all the three RDRP-NSP12 protein for further assessment. The docking protocol implanted in MOE 2016 was first checked for reliability by calculating the (RMSD) root mean square deviation between the crystallized and re-docked conformation of RDRP-NSP12 which was equal to 0.67 Å by using SVL script of MOE, suggesting that the used docking approach is reliable in producing the interaction mode of receptor-ligand complex. We docked 283 hits from ChemBridge database with Ribavirin. A large number of hits were obtained from each database from which top ranked 100 conformations of all docked compounds for each database were saved on the basis of docking score (S).
The resulted binding interactions between these hits and protein were visually observed using protocol implemented in MOE 2016; as a result of refining those molecules which revealed significant interactions with most of the important binding pocket residues (ARG 349, PHE 396, VAL 675, PRO 677, ASN 628 and CYS 395). Finally, 10 best hits were selected from ChemBridge database. The binding energy and binding affinity of top ranked 10 compounds from both databases along with reference compounds were calculated using protocol implemented in MOE. To find out best lead compounds, 3 compounds were selected and their binding energy and binding affinity were calculated as shown in (Table 2).
Table 2:

Binding Modes Study and Selection of Leads Compounds
The 3d ligand interaction and visualization of all the finally identified hits compounds was carried out in order to study their interaction detail. The compound C1 (ChemBridge ID:10509474) showed three stronger hydrogen bonding with active site residues i.e. Arg349, Asn628 and Ser664 showing bond distance, 1.98Å ,2.60Å, and 2.29Å respectively with two hydrophobic interactions with Pro323, Phe326 and Pro667 and Pi interaction with Tyr456. The ligand interaction detail diagram is shown in (Figure 4A). Interaction figure explored that compound C2 (ChemBridge ID: 45091260) forms three Hydrogen bonds with active site residues Pro323, Arg349, and Ser664 with bond distance, 3.71Å, 3.98Å, 3.50Å, and 3.62Å, along with several hydrophobic interactions like, Phe396 and Arg349 as shown in (Figure 4B).C3 (ChemBridge ID:52286894), on the other hand established four hydrogen bond Val675, Arg349, Glu350 and Asn449 also including pie–pie interactions. The interaction pattern of C3 is similar to those of C1, and C2, which also block these residues to disrupt the binding between RDRP-NSP12 as shown in (Figure 4C).
Molecular Dynamics Stability of Complex System
To reveal the stability dynamic properties of the identified hits along with reference drug were examined using a molecular dynamics simulation. The stability of the systems was studied in term of root mean square deviation (RMSD). A total of 100 ns simulation was run to analyze the comparative stability of compound as compared to the reference compound. The graph of RMSD shows that the Ribavirin has deviated more as compared to the five selected complexes which means that selected complexes are more stable as compared to reference complex as shown in (Figure 5). According to simulation results, the RMSD graph of the reference drug ranged from initial point to 6.0. shows maximum instability of the complex (Figure 5). The RMSD value initially rose up to 6 Angstrom, after the completion of 70 ns of MD simulation, but no more fluctuation was not seen after 70 ns, nor from 70 ns to 100 ns of MD simulation, and then more fluctuations were seen from 70-100 ns, indicating that the complex attained its stability after 60 ns. After 100 ns of simulation, the most stable and energy-efficient conformer was obtained; this can be used as a standard (control) for comparison with the generated hits. However, when compared to the reference drug, C1 (ID:10509474), system had the lowest RMSD and no significant fluctuations. The average RMSD for the C1 was 3.1. The RMSD fluctuated slightly between 0 and 100 ns but remained between 2 and 3.9. The C2 (ID:45091260) system had an average RMSD of 1.3, and even while small variations were seen between 0 and 100 ns, the system was typically stable. The C3 (ID:52286894) was stable, however a small fluctuation was seen ranging from 30-60 ns. The stated average RMSD of the C3 system was 3.2. The system became more stable between 70 and 100 ns of molecular dynamic simulation. These results suggested that the identified hits have potential towards the RDRP-NSP12 and bound more rigidly as compared with reference peptide and endorsed the overall stability of the protein complex. All the RMSD graphs are given in (Figure 5).
Figure 4 Ligand interaction of hits compound A. (ChemBridge ID:10509474), B. (ChemBridge ID: 45091260), C. (ChemBridge ID:52286894). The magentas colors show the ligand molecule.

Figure 5 RMSD of the top three hits and the standard compound.

Residues Flexibility Analysis
In order to get more information on how the inhibition quality of these compounds compare among themselves and with the reference compound and how the reference complex protein less reactive root means square fluctuations (RMSF) analysis was performed. Residues flexibility indexing reveals significant information regarding the binding of two proteins, small molecules, molecular recognition, and bioengineering. We calculated the RMSF (root mean square fluctuation) for reference complex and the finally selected hits reported it in (Figure 6). The RMSF plot shows that the residues of each compound in same RDRP (NSP12) protein pocket had entirely different fluctuations in the course of trajectory. The RMSF of each compound in complex were compared with the RMSF plot of reference compound (Ribavirin). The C1 compound i.e. (C1_10509474) showed smaller fluctuations 0.5-2.5 Å of the residues (0-700 amino acids) and 0.5-0.8 Å in the region of residues (710-775 amino acids), smaller fluctuations mean more stable complex. The remaining residues were comparable in fluctuations with the reference compounds, even in some region the reference compound was having fewer fluctuation, but still revising the other analysis like binding energies, docking scores, RMSD as well as RMSF values of C1 seems to be better inhibitor than Ribavirin (Figure 6), The 2nd inhibitor C2 (C2_45091260) has also about same RMSF behavior just like 1st compound having fewer fluctuation of 0.5-2.5 Å in the system in the region of residues (0-700 amino acids) and 0.5-0.8 Å in the region (710-775 amino acids).
Figure 6 RMSF of the top three hits and the standard compound.

The 3rd inhibitor C3 (C3_52286894) has also about same RMSF behavior just like the two one but minor variation C3 compound having fewer fluctuation of 1.0-1.5 Å in the system in the region of residues (10-175 amino acids) and 1.1-1.8 Å in the region (300-400 amino acids). Other regions have equal fluctuation with reference compound and in some region C2, and C1 have lesser fluctuation as compared to the test compound. Although the reference compound seems stable in some regions but still considering other analysis C3 can be better candidate than C1 and C2.
Hydrogen Bond Analysis
The amount of generated hydrogen bonds in all systems was calculated to provide a more precise assessment at the atomic level. The hydrogen-donor-acceptor angle must be 30 degrees, and the donor-acceptor distance must be 0.35 nanometers. When these two conditions are met simultaneously, a hydrogen bond is formed. Hydrogen bonds are necessary for the conservation of secondary structure in peptides and proteins [51,52]. The time-dependent hydrogen bonding investigation revealed that all nine suggested that the final hits had the strongest and best hydrogen-bonding networks with the NSP12 (Figure 7). This was revealed that C3_52286894-NSP12 combination, contain a large amount of hydrogen bonds as compared to the Ribavirin-NSP12. Our findings show, that al the three hits have a high, remarkable NSP12 inhibitors.
Free Energy Calculations
The MM-PBSA/GBSA method is used to compute binding free energy. The molecular mechanical energies and continuous solvent fashions underpin the MMPBSA/ GBSA technique. MMPBSA.py, a Python program, was used to determine the free energy of the reference compound and chosen (searched) compounds. The BFE of the four systems was determined in this study (binding free energy) [53]. The MM/GBSA energies of Ribavirin-NSP12 and the C1_10509474, C2_45091260, and C3_52286894 were calculated. These finding strongly suggest that the retrieved hits involved in the inhibition of NSP12 as compared to Ribavirin-NSP12 complex. The binding free energies for the last 50 ns and their averages were calculated and given in (Table 3). The data shows that reference compound binding free energy (van der Waals energy (-16.6296), C3_52286894 (-53.5816), C2_45091260 (-25.9329), and C1_10509474 (-33.3796) (Table 3), respectively.
Figure 7 H-bond of the top three hits and the standard compound.

Table 3: Binding free energy calculation.

The objective of this study was to perform a virtual screening of compounds from the ChemBridge database, as well as molecular docking and molecular dynamics simulations of selected compounds and a reference ligand (Ribavirin), and to estimate their binding interactions with the RDRP (NSP12) protein of SARS-CoV-2. Three natural compounds, ChemBridge10509474, ChemBridge45091260, and ChemBridge52286894, were found to have strong interactions with the active site of the PqsA enzyme, with binding affinities ranging from -6.2 to -9 kcal/mol. These compounds have improved pharmacophore features compared to the reference compound attached to the RDRP (NSP12) protein. These results suggest that the compounds have the potential to be developed into drugs for the RDRP (NSP12) protein of SARS-CoV-2.

























