 Mini Review
Mini ReviewAbstract
Coronary thrombosis is one of the leading causes of mortality and morbidity in cardiovascular diseases, and patients who received vascular stent treatments are likely to suffer from restenosis due to tissue damage from stenting procedures (extrinsic pathway) and/or presence of unregulated factor XII (intrinsic pathway). Regardless of the pathway, coagulation factors and exposed collagen activate the G-protein-coupled receptors located at the plasma membrane of the resting platelets resulting in the change of their shapes with protrusions of filopodia and lamellipodia for surface adhesion. In this mini review, we discussed the mechanisms involved in platelet activation, adhesion, and aggregation. More importantly, we reviewed the use of polyurethane membranes with modified surface functional groups to down-regulate platelet adhesion and aggregation activities. Polyurethane membranes with hydrophilic and negatively charged surface properties showed a reduced αIIb-β3 signaling from the activated platelets, resulting in the decrease of platelet adhesion and aggregation. The use of polyurethane membranes with modified surface properties as coatings on vascular stents provides an engineering approach to mitigate blood clotting associated with restenosis.
Keywords: Platelets; Activation and Adhesion; Coronary Thrombosis; Blood Clotting; Polyurethane; Surface Properties
Abbreviations: WHO: World Health Organization; PARs: Protease-Activated Receptors; GPCRs: G-Protein-Coupled Receptors; PU-PEG: Polyethylene Glycol- Polyurethane; ELISA: Enzyme-Linked Immunosorbent Assays
Introduction
Cardiovascular diseases account for 31% of all deaths (17.9 million people) worldwide according to the World Health Organization (WHO) [1]. One of the major cardiovascular diseases is coronary thrombosis, and it is associated with the narrowing and/or blockage of coronary arteries that prevent the circulation of blood to the heart tissue resulting in heart attacks and/or heart failures. Development of coronary thrombosis starts with the narrowing of the arteries due to the formation and growth of plaques, consisting of fatty products of cholesterols, calcium, and cellular wastes, inside the vessels wall. When a plaque ruptures, platelets are recruited and accumulated onto the surface of the plaque in order to stop vascular bleeding. The repeating process of plaque rupture and platelet accumulation inside the coronary arteries leads to coronary thrombosis [2]. The current therapeutic methods for this life-threatening cardiovascular disease include balloon angioplasty, vascular stenting, and vascular bypass grafts. However, restenosis, the reoccurrence of narrowed blood vessels associated with vascular tissue damage during angioplasty, can often promote thrombus formation within the first 24 hours that may lead to a second acute heart attack [3]. Placement of vascular stents, including the traditional bare metal stents and the recently developed drug-eluting stents, may cause in-stent restenosis associated with vascular tissue damage during implantation. Furthermore, vascular stents and vascular bypass grafts are considered as foreign objects to the body, which may trigger uncontrollable immune responses that facilitate coagulation. These unregulated enzymatic activities can be categorized into intrinsic (contact) and extrinsic (trauma) pathways that ultimately converge at the generation of factor X (FX) [4]. The generation of activated factor X (FXa) is then catalyzed by activated factor VIII (FVIIIa) and activated factor VII (FVIIa) through the intrinsic and extrinsic pathways, respectively [5]. Local blood clots are formed as FXa interacts with its cofactor FVa resulting in the production of prothrombinase complexes that generate thrombin (FIIa), which promotes the polymerization and deposition of insoluble fibrins. Both the intrinsic and the extrinsic pathways provide signaling proteins that activate platelets to up-regulate their activities in adhesion and aggregation.
In our previous work [4], we have provided a perspective on the applications of engineering membranes, including drug-eluting materials and surface modified polymeric and/or non-polymeric coatings on medical implants to improve hemocompatibility. In particular, our original research work indicated that polyurethane membranes slowed down blood coagulation by 1000-fold as compared to the glass control surfaces [6]. As a continuous effort to investigate the hemocompatibility of polymeric membranes, while providing an overview of current methods performed on the in vitro platelet adhesion assays, we review research works done in the field with modified polyurethane membranes. In this article, we describe the mechanisms involving platelet activation through intrinsic and extrinsic pathways of coagulation followed by narrating how platelets adhere to a surface while recruiting and aggregating more platelets through a positive feedback loop to clot the blood and stop blood loss. This mini review provides research works in the area of polyurethane membranes and its modified surfaces as potential substrates in preventing platelet activation, adhesion and aggregation. The applications of a thin polyurethane coating on the surface of implantable medical devices, such as a vascular stent, provide an engineering approach to safely downregulate coagulation factors that activate platelets to adhere and aggregate into a blood clot.
Platelet Activation and Adhesion
Platelets play a primary role in thrombosis and hemostasis
due to their ability to self-activate in response to signaling cues.
Platelet exposure to collagen and thrombin in an injured vessel due
to extrinsic causes results in the activation of platelets. Through
the intrinsic pathway; however, factors such as surface charges,
hydrophobicity, and topography of a contact surface may activate
platelets and promote their adhesion, aggregation, and activation
[7,8]. In particular, unregulated factor XII (FXII), which is known
to bind to collagen and initiate the coagulation process, promotes
platelet activation. Studies have shown that the FXII-dependent
procoagulant capacity of collagen increased significantly due to the
release of platelet-derived activators such as linker for activation
of T cells (LAT) and phospholipase Cγ2 PLCγ2 [9]. This finding is
further supported by the role of integrin αIIb-β3 signaling from
activated platelets in promoting FXII activation [10]. Irrespective
of the method of activation (e.g., extrinsic or intrinsic causes), these
signaling pathways alter platelets morphology and behavior where
platelets spread more, adhere better, and overexpress filopodia and
lamellipodia for further amplification of the signals [11].
The mechanisms of platelet activation and the subsequent
adhesion and aggregation processes are illustrated in Figure 1
[12]. As indicated, various adhesive Glycoprotein (GP) complexes
on or in the platelets’ plasma membrane interact with extracellular
proteins such as von Willebrand factor (GP Ib-IX) and collagen (GP
VI) [13,14]. In addition, soluble platelet agonists such as thrombin,
serotonin, Adenosine Diphosphate (ADP), and thromboxane A2
(TXA2) are released from the damaged cells as well as the activated
platelets to recruit the circulating platelets via various G-Protein-
Coupled Receptors (GPCRs) [15]. Thrombin activates platelets
through Protease-Activated Receptors (PARs) to stimulate the
approaching platelets [16]. Thrombin also binds with GP Ib-IX
to activate platelets into low-dose thrombin. Serotonin binds
with platelet Gq-coupled receptor, 5HT2A [15]. ADP, reserved in
the granules of platelets, is the metabolic byproduct of cellular
activities, and it activates the resting platelet through purinergic
receptor P2Y12 [17]. TXA2 activates platelets through the TXA2/
prostaglandin H2 receptor (TP) that couples with the GPCRs [18].
Other factors, such as calcium concentration in blood, regulate
the physiologic function of platelets since the increase in calcium
concentration promotes fibrinogen receptor activation [19].
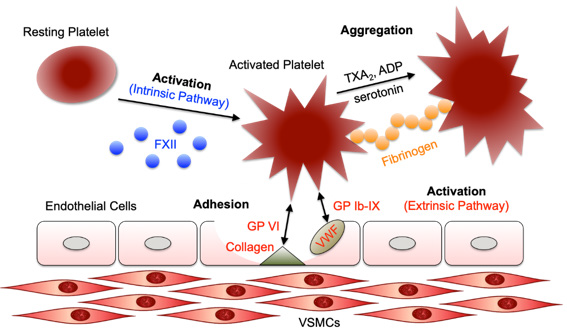
Figure 1: A schematic illustration on the mechanisms of platelet activation via intrinsic and/or extrinsic causes followed by platelet adhesion to a surface and the sequential signaling events in aggregation. Figure is redrawn from [12].
Platelet adhesion and activation due to restenosis is of particular concern, especially for the design of engineering surfaces that inhibit blood clotting from platelet aggregation. For example, it has been shown that the negatively charged surfaces not only down-regulate the conversion of the adsorbed fibrinogen to fibrin [7], which impedes the coagulation process, but also reduce platelet aggregation through mechanisms of passive agglutination (platelets are negatively charged) and integrin αIIbβ3-mediated aggregation [20]. In addition, increasing surface hydrophobicity significantly increases fibrinogen adsorption, which promotes platelet adhesion [21]. However, hydrophobic surfaces that exhibit contact angles greater than 120 degrees show better blood compatibility and less potential for platelet adhesion and activation [22]. Finally, a 2.5-fold reduction on platelet aggregation suggests the effects of surface topography (i.e., from 700 nm to 400 nm) on bulk platelet activation [23]. In general, platelet activation and adhesion through intrinsic pathway of surface contacts of foreign objects are related to various cellular mechanisms on the activation of protein receptors as well as surface physicochemical properties of the implantable devices (e.g., vascular stents).
Engineering Surfaces That Prevent Platelet Adhesion
intensive research efforts. Providing an engineering solution to
prevent platelet adhesion and subsequent activation by implantable
devices, such as vascular stents, is one such effort. Even though drugs
such as aspirin are a potential preventive solution to restenosis,
platelet aggregation on the other hand is a natural process that is
necessary to maintain proper functioning of the body, especially
when injury occurs [24]. To locally down-regulate platelet
activation and adhesion ability while still being able to maintain
its function in coagulation process, surface modifications of the
implantable devices become an ideal engineering solution. This
approach leads to the investigation of the platelet compatibility on
Polyethylene Glycol–Polyurethane (PU-PEG) coatings. Implantable
devices in contact with blood in current implementations have
short lifespans predominately due to blood coagulation. Studies
involving polymer coatings with modified surface functional groups
have been conducted in an effort to provide a permanent solution
for a hemocompatible device. A recent in vitro study indicated that
the Polyethylene Glycol (PEG) modified polyurethane (PU) surfaces
with different terminal groups (–OH, –NH2, and –SO3) prevents
platelet activation and adhesion [25].
Results showed 1.3-fold and 1.7-fold increases of free calcium
concentration in the PEG-modified PU surfaced terminated with
hydroxyl and amine functional groups, respectively, as compared to
the PEG-modified PU control groups after 20 minutes of incubation.
The higher free calcium concentration in the PU surfaces terminated
with amine functional groups was due to the ionic interactions
between the amine and the platelet membrane. In addition, the
sulfonate functionalized PEG-modified PU surfaces showed a 0.4-fold decrease in free calcium concentration as compared to the PU
control groups, suggesting the dependence of platelet activation
on surface functional groups of polymer coatings. In vitro platelet
adhesion assays were performed on these surfaces, and the PEGmodified
PU surfaces showed 47% less platelet adhesion than the
blank PU controls after 10 minutes of incubation. Platelet adhesions
decreased to 27%, 21%, and 12% on the PEG-modified PU surfaces
terminated with hydroxyl, amine, and sulfonate functional groups,
respectively. These findings demonstrate the effects of surface
functional groups on the inhibition of platelet activation and
adhesion.
In addition to the surface chemical nature and ionic charges,
surface wettability of PU coatings can be used as an effective surface
modifier [6]. In a study, soybean-derived Phosphatidylcholine (PC)
modified PU coatings were prepared by dipping PU coatings in PC
containing PU solutions followed by solvent evaporation [26]. The
resulting PC-modified PU coatings exhibited an increase in surface
wettability indicated by contact angle measurements. The PCmodified
PU surfaces consisted of phosphorylcholine end groups
on the coating surface, mimicking the physiologic endothelial
membrane structure that significantly reduced the adsorption
of plasma-derived proteins, such as fibrinogen, fibronectin, or
von Willebrand factor. Enzyme-Linked Immunosorbent Assays
(ELISA) compared the surface absorptions of fibrinogen in platelet
poor plasma on the PC-modified PU to that of PU alone. The data
showed a greater than 3-fold reduction in the amount of adsorbed
fibrinogen on the PC-modified PU groups than the PU controls.
Furthermore, hemocompatibility analysis under dynamic shear
stress testing conditions using a platelet analyzer suggested that
more than 70% of platelets were maintained in the blood samples
after contacting the PC-modified PU surfaces.
Effects of polymer swelling ability have a direct influence on
the physical cues of platelet adhesion. Different substituents at
the N-position of poly(N-alkyl acrylamide) coatings were dipcoated
onto PU substrates to investigate the swelling ability
(hydrophilicity) of the coatings on the platelet adhesion [27].
These coatings, irrespective to the number and length of alkyl
substituents at the N-position that attributed to varying swelling
degree, exhibited little to negligible platelet adhesion due to the
bioinertness of poly(N-alkyl acrylamides) that repelled proteins
and endothelial cells. Furthermore, crosslinking of the poly(Nalkyl
acrylamide) reduced the swelling ability and improved
the mechanical properties of the coating under shear stress
environment without affecting the thrombogenic properties of the
surfaces by preventing platelet adhesion.
Surface modifications of the PU membranes with a small
molecule antiplatelet drug, dipyridamole, demonstrated the abiity
to reduce platelet adhesion. In a study, dipyridamole was used in
two variations on the surface modification of the PU membranes
[28]. The two groups included linking dipyridamole directly to PU membranes and linking dipyridamole to PU membranes with
a short hydrophilic spacer chain. After 15, 30, and 60 minutes of
incubation in human platelet rich plasma, platelet adhesion density
of the dipyridamole-linked PU membranes decreased 72%, 35%,
and 52% as compared to the PU controls, respectively. In addition,
the platelet adhesion density from the dipyridamole-linked PU
membranes with hydrophilic spacers decreased 63%, 28%, and
18% as compared to the PU controls, respectively. Interestingly, at
short incubation time (e.g., 15 min), PU membranes grafted with
only the hydrophilic spacers increased the platelet adhesion density
by 130%. This trend was further decreased to 53% at 30 minutes
and 27% at 60 minutes, suggesting that hydrophilic surfaces were
more hemocompatible perhaps due to the mechanisms involved in
platelet spreading and aggregation.
Incorporating Polyethylenimine (PEI) in PU membranes
enhances the surface grafting sites of primary amine (–NH2).
Tethering of heparin and phosphorylcholine (PC) groups on the
surface of PEI-PU membranes inhibited platelet adhesion [29].
Water uptake tests demonstrated that blank PEI-PU membranes,
heparin-tethered PU membranes, and PC-grafted PU membranes
were 23.6x, 13.6x, and 15.9x more hydrophilic than the PU control
groups, respectively. The low water contact angles (35°~40°) of
these modified surfaces supported the water uptake data and
showed that the surfaces of these membranes were hydrophilic.
Static platelet adhesion assays using platelet rich plasma on PU
control groups showed platelet aggregates, where the adhered
platelets exhibited long pseudopods, which is evidence of platelet
activation to spread over the surfaces. Platelet adhesion on PEI-PU
membranes were reduced by 48%, whereas both heparin-tethered
PU membranes and PC-grafted PU membranes demonstrated 1000-
fold decreases in platelet adhesion.
Taken together and summarized in Table 1 [25-29], the surface
modified PU membranes demonstrate the promising ability in
inhibiting platelet activation and adhesion. These surfaces are
either hydrophilic or swellable, which repel the proteins deposition
during the signaling events of platelet adhesion and aggregation.
In addition, surface modifications of PU membranes allow grafting
of antiplatelet molecules to prevent the deposition of plates on the
coating surfaces. These techniques provide a much-improved life
span on the implantable medical devices, such as vascular stents.
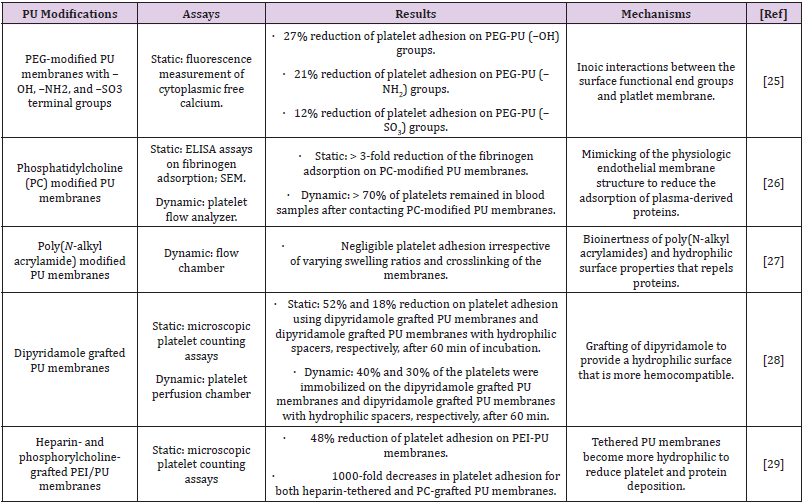
Table 1: Summary of thrombogenicity of modified Polyurethane (PU) surfaces.
Conclusion and Future Directions
play a fundamental role in clot formation and
preventing hemorrhage, a necessary and protective function in
hemostasis. Thrombosis, however, is a pathologic state when
unwanted blood clots are formed inside the arteries. Thrombosis
may occur through the extrinsic (trauma) or intrinsic (contact) pathways, and both pathways converge into the formation of
thrombin that activates the polymerization and deposition of fibrin.
Each pathway provides important signaling factors to activate
platelets via the G-protein-coupled receptors located at the plasma
membrane. Once activated, filopodia and lamellipodia protrusions
provide platelets with adhesion ability to the surface. Crosstalks
between the activated platelets enable the aggregation of platelets
into a blood clot.
Several polyurethane membranes with modified surface
properties, either by promoting the surface wettability or
enhancing the surface negative charge, have shown promising
results in preventing platelet adhesion or aggregation. These types
of thin coatings may be used on implantable medical devices (i.e.,
vascular stents) to improve their therapeutic functions in the
body by reducing the chances of having a restenosis. The future of
polymer coatings may facilitate the development of multi-functional
membranes, such as a composite, to provide active, as well as
passive, protections to the stents. In addition, surface grafting of
anti-clotting functional groups tailored to patients’ needs is also a
promising research topic in anticoagulant polymer membranes.
Acknowledgment
This work was supported by a grant from the American Heart Association (18AIREA33960372) awarded to S.F.C.
Author Contribution
All authors contributed equally on the manuscript. In particular, S.F.C and B.A.C. contributed to the writing of the original manuscript; A.A. and P.F.N. contributed to the critical review and proofreading of the manuscript; S.F.C and P.F.N contributed to funding acquisition; S.F.C., A.A., and P.F.N contributed to the project administration.
Conflict of interest
The authors declare no conflict of interest regarding the publication of this paper
References
- World Health Organization (2017) Cardiovascular diseases (CVDs). World Health Organ.
- Badimon L, Padró T, Vilahur G (2012) Atherosclerosis, platelets and thrombosis in acute ischaemic heart disease. Eur Heart J Acute Cardiovasc Care 1(1): 60-74.
- Hamid H, Coltart J (2007) ‘Miracle stents’ - a future without restenosis. McGill J Med MJM Int Forum Adv Med Sci Stud 10(2): 105-111.
- Wilson AC, Neuenschwander PF, Chou S F (2019) Engineering approaches to prevent blood clotting from medical implants. Arch Biomed Eng Biotechnol 1(2): 5.
- Grover SP, Mackman N (2018) Tissue factor: An essential mediator of hemostasis and trigger of thrombosis. Arterioscler Thromb Vasc Biol 38(4): 709-725.
- Wilson AC, Chou S F, Lozano R, Jonathan Y Chen, Pierre F N (2019) Thermal and physico-mechanical characterizations of thromboresistant polyurethane films. Bioengineering 6(3): 69.
- Evans Nguyen KM, Schoenfisch MH (2005) Fibrin proliferation at model surfaces: Influence of surface properties. Langmuir 21(5): 1691-1694.
- Koc Y, De Mello AJ, Mc Hale G, M Newton, P Roach, et al. (2008) Nano-scale superhydrophobicity: Suppression of protein adsorption and promotion of flow-induced detachment. Lab Chip 8: 582-586.
- Van der Meijden PEJ, Munnix ICA, Auger JM, José W P Govers Riemslag, Judith M E M Cosemans et al. (2009) Dual role of collagen in factor XII–dependent thrombus formation. Blood 114(4): 881–890.
- Huang J, Li X, Shi X, Mark Zhu, Jinghan Wang, et al. (2019) Platelet integrin αIIbβ3: signal transduction, regulation, and its therapeutic targeting. J Hematol OncolJ Hematol Oncol 12(1): 26.
- Bearer EL, Prakash JM, Li Z (2002) Actin dynamics in platelets. International Review of Cytology, Elsevier 217: 137-182.
- De Abajo FJ (2011) Effects of selective serotonin reuptake inhibitors on platelet function: Mechanisms, clinical outcomes and implications for use in elderly patients. Drugs Aging 28(5): 345-367.
- Clemetson KJ, Clemetson JM (2001) Platelet collagen receptors. Thromb Haemost 86(1): 189-197.
- Du X (2007) Signaling and regulation of the platelet glycoprotein Ib–IX–V complex. Curr Opin Hematol 14(3): 262-269.
- Offermanns S (2006) Activation of platelet function through G protein–coupled receptors. Circ Res 99(12): 1293-1304.
- Coughlin SR (1999) How the protease thrombin talks to cells. Proc Natl Acad Sci 96(20): 11023-11027.
- Ohlmann P, Laugwitz KL, Nürnberg B, K Spicher, G Schultz, et al. (1995) The human platelet ADP receptor activates G i2 proteins. Biochem J 312(3): 775-779.
- Knezevic I, Borg C (1993) Identification of Gq, as one of the G-proteins which copurify with human platelet thromboxane A2/prostaglandin H2 receptors. J Biol Chem 268(34): 26011-26017.
- Quinton TM, Ozdener F, Dangelmaier C, James L Daniel, Satya P Kunapuli (2002) Glycoprotein VI–mediated platelet fibrinogen receptor activation occurs through calcium-sensitive and PKC-sensitive pathways without a requirement for secreted ADP. Blood 99(9): 3228-3234.
- Zia F, Kendall M, Watson SP, Paula M Mendes (2018) Platelet aggregation induced by polystyrene and platinum nanoparticles is dependent on surface area. RSC Adv 8(66): 37789-37794.
- Fischer M, Sperling C, Werner C (2010) Synergistic effect of hydrophobic and anionic surface groups triggers blood coagulation in vitro. J Mater Sci Mater Med 21(3): 931-937.
- Massa TM, Yang ML, Ho JYC, J L Brash, J P Santerre (2005) Fibrinogen surface distribution correlates to platelet adhesion pattern on fluorinated surface-modified polyetherurethane. Biomaterials 26(35): 7367-7376.
- Milner KR, Siedlecki CA, Snyder AJ (2005) Development of novel submicron textured polyether(urethane urea) for decreasing platelet adhesion. ASAIO J 51(5): 578-584.
- Roth GJ, Majerus PW (1975) The mechanism of the effect of aspirin on human platelets. I. Acetylation of a particulate fraction protein. J Clin Invest 56(3): 624-632.
- Park KD, Suzuki K, Lee WK, J E Lee, Y H Kim, et al. (1996) Platelet adhesion and activation on polyethylene glycol modified polyurethane surfaces: Measurement of cytoplasmic calcium. ASAIO J 42(5): M876-M881.
- Butruk Raszeja B, Trzaskowski M, Ciach T (2015) Cell membrane-mimicking coating for blood-contacting polyurethanes. J Biomater Appl 29(6): 801-812.
- Baghai M, Tamura N, Beyersdorf F, Michael Henze, Oswald Prucker, et al. (2014) Platelet repellent properties of hydrogel coatings on polyurethane-coated glass surfaces. ASAIO J 60(5): 587-593.
- Aldenhoff Y, Koole L (2003) Platelet adhesion studies on dipyridamole coated polyurethane surfaces. Eur Cell Mater 5: 61-67.
- Tan M, Feng Y, Wang H, Musammir Khan, Jintang Guo, et al. (2013) Immobilized bioactive agents onto polyurethane surface with heparin and phosphorylcholine group. Macromol Res 21: 541-549.
