 Short Communication
Short CommunicationAbstract
Background: Bullous Congenital Ichthyosiform Erythroderma (BCIE) is a rare form of congenital ichthyosis which is characterized by hyperkeratosis with or without scaling [1]. Otological manifestations typically result occur due to the deposition and impaction of abnormal keratin in the external auditory canal causing conductive hearing loss and increased susceptibility to ear infections [2].
Methods: A retrospective study of patients who presented to our service between 2003 and 2013 was completed examining incidence, presentation and management of otological manifestations of congenital icythosis.
Results: 6 patients were identified: 66% (n=4) were male and 33% (n=2) were female. The age at first presentation ranged from 5 months– 4 years. 100% of patients (n=6) presented with conductive hearing loss with no preceding infection or otorrhea. Examination under general anesthesia (EUA) revealed ear canal keratin plugs and the absence of middle ear disease. Management was conservative consisting of regular application of olive oil drops and repeated microdebridement.
Discussion and Conclusion: Few reports have addressed the otological manifestations of BCIE and more work is needed to investigate this area. Our outcome provides additional insight demonstrating that conservative management with the regular instillation of olive oil drops and repeated EUA and microdebridement of keratin plugs to prevent complications.
Abbreviations: MER: Middle Ear Reflexes; OAE: Otoacoustic Emission; OPD: Outpatient Department; SNHL: Sensorineural Hearing Loss
Short Communication
BCIE, also known as Epidermolytic Ichthyosis is a rare form of
Congenital Ichthyosis. Jean Louis Brocq first described ‘erythrodermie
congenitale ichthyosiforme avec hyperepidermotrophie’
in 1902 [3]. The incidence is about 1: 200,000 with most cases
presenting shortly after birth [4]. The pathogenesis of the condition
involves mutations of the genes encoding KRT1 and KRT
10 affect the structural integrity of the suprabasilar layers of the
epidermis [5-8]. This results in defective keratin formation and
disturbance of the keratin cytoskeleton. Mutations lead to abnormal
keratin filament clumping resulting in skin collapse and fragility.
BCIE is a lifelong condition characterized by marked skin
fragility with blistering and erythroderma at birth. The greatest
risk to life is in the neonatal period when neonates are susceptible
to dehydration and sepsis. Management at this point
aims to minimize trans epidermal water loss, support caloric needs
and prevent complications.
Although an amelioration of symptoms can occur, an ichthyotic
skin disorder persists throughout the lifetime of the patient.
The spectrum of disease can be quite disfiguring, depending on
the severity, type and extent of the body surface area. The head,
neck, ears and extremities are typically involved, and symptoms
include skin discomfort, xerosis, pruritus, hypo- or anhidrosis/
heat intolerance, vision and ophthmologic abnormalities. There
is no cure at present and treatment is symptomatic consisting
of various topical emollients, keratolytics and medications,
and sometimes systemic retinoids [9]. There is a paucity of
research on the otological manifestations of the disease and
exact incidence remains unknown. The clinical manifestations of severe keratinization can affect the pinna and external auditory
canal resulting in the impaction of the EAC with thick plaques.
This predisposes patients to fungal and bacterial infections and
conductive hearing loss. Otological phenomenon of the condition
can present a management conundrum to otolaryngologists, as
there are currently no guidelines to assist in making decisions
regarding optimum management.
Methods
A retrospective observational study was carried out of patients presenting to the Department of Otorhinolaryngology, University Hospital Limerick. Patient medical records were reviewed, and the following information was sought patient demographics, relevant family history, clinical findings, type of hearing loss, results of diagnostics and therapeutic interventions.
Results
Six patients were identified: 66% (n=4) were male and 33% (n=2) were female. The female patients were twin sisters. Initial age at presentation ranged from 5 months to 4 years. 2 patients presented with X-linked ichthyosis. All patients had similar clinical presentations and clinical findings: hearing loss with no history of otorrhea or ear infections. Type B tympanograms and hearing loss was recorded. Otoacoustic emission (OAE) and middle ear reflexes (MER) were within normal range in all patients post procedure. All patients were booked for examination under general anesthesia (EAU). This revealed ear canal keratin plugs with some inflammatory skin changes in the absence of infective otitis externa or middle ear disease (Figures 1-3). Post operatively patients were reviewed in the outpatient department (OPD) at six weeks. If early ear canal keratinisation was observed at this time patients were commenced on olive oil ear drops and were re-scheduled for a second EUA and microdebridement of ears at a three-month interval. Preoperative tympanograms were type B. EUA results were similar demonstrating keratin plugs which underwent microdebridement. Post-operative tympanogram were within normal ranges.
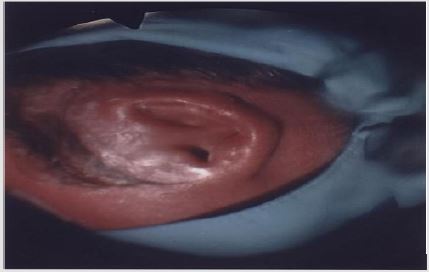
Figure 1: Image of patients Pinna at EUA.
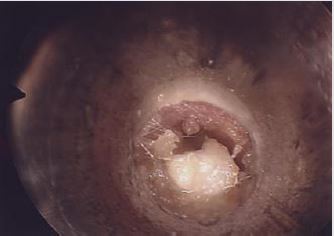
Figure 2: Keratin plug in external ear canal at EUA.
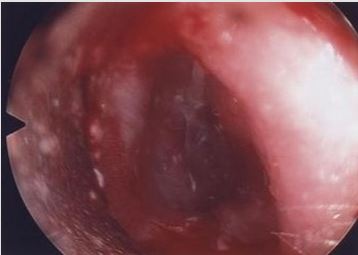
Figure 3: Patient’s ear canal following microdebridement.
A third surveillance EUA was scheduled depending on the severity and observed re-accumulation after a 6-month period while patients were continued on olive oil drops and similar clinical and audiological findings were present as on previous assessment. An individualized time interval for the following EUA was extended and rescheduled at a 9 months period with continuous regular olive oil ear drops instillation. Similar clinical and audiological finding were found at follow-up. Accumulations recurred quite quickly but persistent symptoms did not develop for approximately 9 months and were proportional to the severity of the generalized condition. Assessments were repeated at a 9-12 monthly interval either in the clinic or operating theatre depending on age and tolerance for each patient, unless early procedures were deemed clinically necessary or the intervals could be extended out further. In total patients had on average 3 EUAs with microdebridement while as they got older the condition could be managed in the clinic with increasingly less frequent visits. From our cohort of patients 16% (n=1) had evidence of infective otitis externa in some but not all of his EUAs.
Discussion and Conclusion
The overall management of Congenital Ichthyosis is complex and in the absence of definitive curative management supportive care is offered to reduce symptoms and prevent complications. Topical emollients and ketalolytics are the mainstay of treatment [10]. More aggressive management strategies include the institution of retinoid therapies but there is understandable reluctance to do given their adverse effects. It is postulated that gene therapy may provide therapeutic solutions in the future. There are few references to the otological manifestations of Congenital Icythyosis in the literature. One prospective Spanish study published in 2015 examined ear involvement in non-syndromic autosomal recessive congenital ichthyosis. Unselected random patients were enrolled as a snapshot survey from a Dermatology clinic and 100% of the study population had otological manifestations, 37.5% reported a history of ear pain, pruritis or otitis media and 25% were found to have conductive hearing loss. Interestingly none of the patients had been referred to ENT previously and none had been prescribed topical ear treatments at any stage. With adequate ear care, cleaning and unblocking 50% regained hearing. This was the only trial that examined the otological manifestations but was certainly limited by the small population number. Importantly though it further strengths the management strategy our group proposes.
It is extremely important when assessing these patients to accurately determine the type of hearing loss they have as different subtypes of ichthyosis are linked with specific types of hearing loss. BCIE is commonly associated with CHL secondary to EAC keratin plugs. Anther subgroup of this condition such as Keratitis Icythyosis Deafness Syndrome is associated with sensorineural hearing loss (SNHL) [11]. The severity of the SNHL varies depending on the gene type implicated in the mutation which can co-assemble in the cochlear gap junctions [12]. Early recognition of this type of gene mutation related SNHL is essential for consideration of cochlear implants to prevent the development of deaf mutism [13]. Other rare cases of a mixed type hearing loss secondary to vertical gene mutations were also reported in the literature [14]. While the general management of CBIE is complex, management of otological manifestation is relatively simple consisting of regular use of olive oil drops to prevent accumulation and facilitate the removal of cerumen and keratin with regular micro debridement. There are no guidelines or management protocol available for the otological manifestations of this condition. Our management plan was simple with satisfactory outcomes. This consisted of early assessment of symptomatic patients with BCIE with preoperative hearing screening, early EUA with microdebridement of keratin and followed by full audiological assessment.
If middle ear disease, SNHL and/or mixed type hearing loss is evident then patients would have had repeated genetic typing and underwent CT assessment of their temporal bones to exclude other middle or inner ear pathology. The management of patients with normal middle ear whose only complaint is CHL secondary to keratin plugs was simple including the regular use of olive oil drops and easy access to EUA and microdebridement when needed. We found that the frequency of toilet was related to the extent the disease and that as patients got older the need for general anesthesia lessened as did the frequency of clinic visits. We did not encounter any patient with expansile or erosive changes from the disease. Our group suggests using this algorithm to manage these patients although describing a small number of patients is still to our knowledge the largest case series with longest follow-up of this condition to date that we could find in the English literature. While there are no current guidelines in this area our experience is mirrored by case reports describing the management of this condition. Few reports have addressed the otological manifestations of BCIE and more work is needed to provide clarification in a management protocol of a rare process and deliver a prognostic estimate to the parents or attending physicians. Our outcome provides additional insight demonstrating that conservative management with the regular instillation of olive oil drops and repeated EUA and microdebridement of keratin plugs to prevent complications. No ethical approval was required for the study.
References
- Paller AS, Mancini AJ (2011) Hereditary disorders of cornification. Hurwitz clinical pediatric dermatology, (4th), Elsevier, Edinburgh, UK, p. 92-114.
- Diaz LZ, Browning JC, Smidt AC, Rizzo WB, Levy ML (2013) Complications of ichthyosis beyond the skin. Dermatol Ther 26(1): 39-45.
- Thomas Bogenrieder, Michael Landthaler, Wilhelm Stolz (2003) Bullous Congenital Ichthyosiform Erythroderma: Safe and Effective Topical Treatment with Calcipotriol Ointment in a Child. Acta Dermato Venereologica 83(1): 52-54.
- Di Giovanna JJ, Bale SJ (1994) Clinical heterogeneity in epidermolytic hyperkeratosis. Arch Dermatol 130: 1026-1035.
- Cheng Jian, Syder AJ, Yu QC, Letai A, Pallar AS, et al. (1992) The genetic basis of epidermolytic hyperkeratosis: A disorder of differentiation-specific epidermal keratin genes. Cell 70(5): 811-819.
- Chipev CC, Korge BP, Markova N, Bale SJ, DiGiovanna JJ, et al. (1992) A leucine----proline mutation in the H1 subdomain of keratin 1 causes epidermolytic hyperkeratosis. Cell 70(5): 821-828.
- Rothnagel JA, Dominey AM, Dempsey LD, Longley MA, Greenhalgh DA, et al. (1992) Mutations in the rod domains of keratins 1 and 10 in epidermolytic hyperkeratosis. Science 257(5073): 1128-1130.
- Yang JM, Chipev CC, DiGiovanna JJ, Bale SJ, Marekov LN, et al. (1994) Mutations in the H1 and 1A domains in the keratin 1 gene in epidermolytic hyperkeratosis. J Invest Dermatol 102(1): 17-23.
- Lucia Z Diaz, John C Browning, Aimee C Smidt, William B Rizzo (2013) complications of ichthyosis beyond the skin Dermatological Therapy 26(1): 39-45.
- JJ Di Giovanna, T Mauro, LM Milstone, M Schmuth, JR Toro (2013) Systemic retinoids in the management of ichthyoses and related skin types. Dermatologic Therapy 26(1): 26-38.
- Simon Angeli, Xi Lin, Xuezhong Liu (2012) Genetics of Hearing and Deafness. The Anatomical Record 295(11): 1812-1829.
- Liu Xz, Yuan Y, Yan D, Emilie Hong Ding, Xiao Mei Ouyang, et al. (2009) Digenic inheritance of non-syndromic deafness caused by mutations at the gap junction proteins Cx26 and Cx31. Hum Genet 125(1): 53-62.
- EJ Barker, R JS Briggs (2009) Cochlear implantation in children with keratitis-ichthyosis-deafness (KID) syndrome: Outcomes in three cases. Cochlear Implants Int 10(3): 166-173.
- B Kelly, A Lozano, G Altenberg, T Makishima (2008) Connexin 26 mutation in Keratitis-ichthyosis-deafness (KID) syndrome in mother and daughter with combined conductive and sensorineural hearing loss. International Journal of dermatology 47(5): 443-447.
