 Review Article
Review ArticleAbstract
Anti-Neutrophil Cytoplasmic Autoantibody (ANCA)-Associated Vasculitides (AAVs) are rare disorders characterized by inflammation of the small blood vessels resulting in ischemia or haemorrhage. The main phenotypes of AAVs are Granulomatosis with Polyangiitis (GPA), Microscopic Polyangiitis (MPA), and Eosinophilic Granulomatosis with Polyangiitis (EGPA). Although the pathogenesis of these disorders is still incompletely understood, distinct roles for T cells subsets and ANCA autoantibodies were underscored. AAVs present in the head, neck, lung, kidney, skin, heart, eyes, and intestine. The treatment of AAVs aims to induce and maintain remission and rapidly control of the disease activity. Rituximab, the first major alternative to cyclophosphamide plus glucocorticoids therapy, has enhanced the management of AAVs. However, continuous evaluation is constantly needed to manage the uncommon clinical features which may accompany the disease. This review summarizes the major findings of recent studies related to the three kinds of AAVs focusing on new novelties of their pathogenesis, clinical manifestations, and management.
Abbreviations: ANCA: Anti-Neutrophil Cytoplasmic Autoantibody; AAVs: ANCAAssociated Vasculitis; GPA: Granulomatosis with Polyangiitis; EGPA: Eosinophilic Granulomatosis with Polyangiitis; MPA: Microscopic Polyangiitis; CRP: C-Reactive Protein
Introduction
Vasculitides refer to a group of pathological conditions resulting from inflammation of blood vessels, with consequent adverse effects such as ischemia and hemorrhage. According to vessels size, they are usually classified into three groups; small, medium, and large vessel vasculitides. Granulomatous Polyangiitis (GPA), Microscopic Polyangiitis (MPA), and Eosinophilic Granulomatosis with Polyangiitis (EGPA) are the main AAVs types which usually affect small-vessels and characterized by the presence of ANCA autoantibodies in patient’s serum [1,2]. Although they are idiopathic, their pathogenesis and management still a hot topic [3,4]. Divers factors were involved in AAVs pathogenesis including, T cells and complement system activation, and genetic and environmental factors [5]. The details of AAVs pathogenesis are incompletely unexplained. However, evidence related to patients with AAVs suggests a role for macrophage or monocyte in increasing the intensity of tissue injury. In addition, serum of AAVs patients characterized by increased levels of colony stimulating factor 1 (CSF1) [6]. Johansson ÅC et al. found that AAVs pathogenesis was amplified through phagocytes behaviors impairment [7]. ANCApositive glomerulonephritis is may seem to be accompanied with increased C-Reactive Protein (CRP), an acute-phase protein of inflammation related to interleukin 6 (IL-6). Moreover, of primary contributors in AAVs pathogenesis are the leukocytes via damage caused by their migration and invasion. AAVs genetic background knowledge is increased by additional genome-wide association study (GWAS) results and other genomic approaches [8].
In diagnosis, the common laboratory tools in AAVs diagnosis are indirect Immunofluorescence test (IFT) for ANCA screening and then confirm positive results by enzyme-linked immunosorbent assay (ELISA) to differentiate proteinase 3 (PR3)-ANCA or c-ANCA from myeloperoxidase (MPO)-ANCA or p-ANCA (Figure 1). In treatment, therapeutic strategies have been established for AAVs associated with increasing evidence related to their pathogenesis. These strategies aim to prevent disease relapse, reduce drugs toxicity, and limit disease consequences [9]. Two phases are considered in AAVs treatment, the first is an induction of remission, and the second is for rapid control of disease activity [10]. Currently, the monoclonal antibodies, rituximab, and mepolizumab have been reported as novel drugs in AAVs treatment [11,12]. Our previous data confirmed that TNF-α blockers in rheumatic diseases e. g. rheumatoid arthritis compromised the disease activity [13] whereas, TNF-α blocker, etanercept characterized by increasing solid malignancies development [14].
The majority of patients with AAVs develop new cascades of events led to new severe symptoms. Thus, Grayson and his co-workers suggested that continuous evaluation of patients with established vasculitis remains critical [15]. AAVs research directed towards investigating the disease duration optimization and maintenance therapy frequency as well as targeted treatment development which could unravel successful treatment or cure for AAVs. In this review, we focused on the outputs of current studies related to the main three kinds of AAVs, and prospects of alternative treatments or cure.
Granulomatosis with Polyangiitis (GPA)
GPA is a rheumatic systemic small vessels inflammation that affects multiple organs and usually accompany with c-ANCA or rarely p-ANCA autoantibodies. The most predominantly affected population category is age bracket of 40 - 55 years. The disease is frequently presented in a sino-nasal cavity, kidney, and the lung (Figure 1). Klinger in the year 1931 characterized GPA for the first time, then Friedrich Wegener, hence the name Wegener’s granulomatosis was introduced by Godman and Churg 1954 [16- 19]. Even though the etiology of GPA is still unknown, stimulation of TLR2 and TLR9, and the priming effect of TLR ligands on PMNs contributed to GPA pathogenesis by increase proteinase 3 (mPR3) expressions of membrane origin [20]. Also, IL-32, tartrate-resistantacid- phosphatase (TRAP), multinucleated giant cells (MNGs), programmed death receptor-1 (PD-1) and mitochondrial DNA played roles in GPA pathogenesis [21-23]. Moreover, GPA pathogenesis was augmented by upregulation of platelets neutrophils extra-cellular traps (NETs) depend on elevated myeloperoxidase-DNA complexes and platelet-neutrophil aggregates [24]. Meanwhile, α1-antitrypsin deficiency and levamisole are reported as GPA inducer [25,26].
GPA Clinical Features and Management: a new Progress
GPA clinical manifestations were summarized in Table 1. The most common sites involved in GPA are ear, nose, and throat (ENT); lungs; and kidneys (ELK) (Figure 1). Recently, hearing loss, toothache, oral ulcer, rash, otitis media, gastrointestinal complications, ischemic stroke, bilateral ulcers on the face, and fever have been observed in GPA patients [27,35]. The clinical manifestations of GPA in children and adults’ patients were similar except organ involvement frequencies [29]. Skin and gastrointestinal manifestations were considered as markers for GPA severity [27,30]. Despite neurological manifestation is not uncommon with GPA, CNS clinical features in GPA could cause fatal events. Hypertrophic pachy meningitis with unchanged PR3-ANCA level was reported as a unique consequence of GPA [31,32]. Because of central and peripheral nervous system involvement, inpatients with neurologic disorders are more susceptible to GPA [33]. In rare cases, the skull base could be injured in patients with GPA [34]. Also, GPA was associated with chronic lymphocytic leukemia, dengue fever, acute onset progressive dyspnea, and Crohn’s disease [3,36-39].
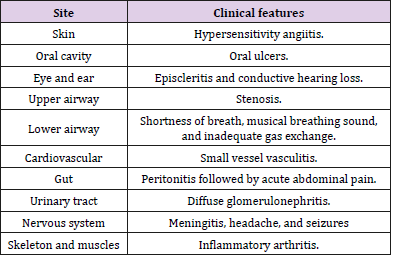
Table 1: GPA clinical symptoms (35).
On the other hand, the cardiac involvement in GPA was rare and presented as coronary vasculitis, myocarditis, or pericarditis [40]. Of cardiac involvement, valvulopathy, bacterial endocarditis, and acute aortic and mitral valve perforations were reported as secondary features for GPA [41-43]. Meanwhile, GPA misdiagnosed as an infective endocarditis in adolescent male [31]. The traditional drugs for GPA treatment are cyclophosphamide and glucocorticoids. However, recent studies have shown that patients with severe AAVs were successfully managed by rituximab [11,47,48]. Abatacept also achieved good results in treating patients with GPA [44]. In the past, the management of GPA-associated peripheral ulcerative keratitis (PUK) was a burden and lacked exact treatment protocol, but now it was controlled relatively by rituximab [45]. Meanwhile, GPA case with refractory necrotizing scleritis associated with PUK was successfully treated by surgical intervention [46]. It has been described that rituximab was effective in GPA with heart involvement [47], but low immunoglobulin levels in patients on rituximab increase the risk of severe hypogammaglobulinemia [48].
As well, the first case of GPA presented as gastric ulcer was successfully treated with rituximab [49]. In addition, GPA association with frosted branch angiitis was improved by intravenous solumedrol of high-dose therapy [50]. Also, it has been suggested that early diagnosis, immunosuppressive therapy, and facial vascularized composite allotransplantation could manage a rare association between malignant pyoderma and GPA [51]. Indeed, Subglottic Stenosis (SGS) in GPA may be relieved by subglottic dilatation, but the patient will still at recurrence risk. As well, tracheostomy is recommended to manage severe airway-limiting stenosis [52]. A recent report suggested that differential diagnosis should include hypersensitivity pneumonitis when a GPA patient in azathioprine has fresh pulmonary lesions [53]. On the other hand, receiving maintenance immunosuppressive therapy in a patient with GPA or GPA associated with systemic lupus erythematosus could provoke acute kidney injury or alveolar hemorrhage [54,55].
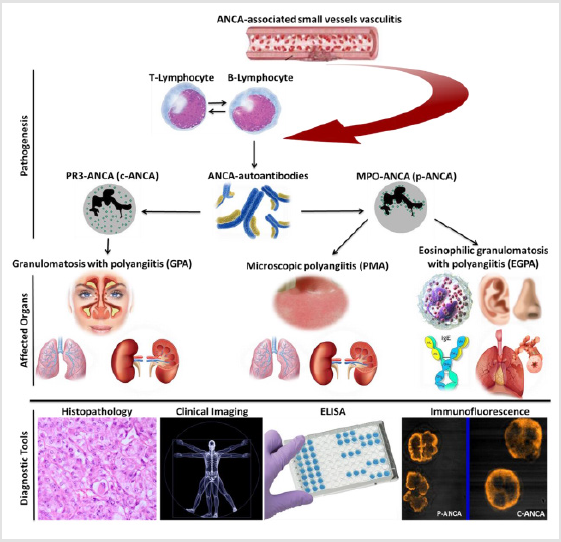
Figure 1: An overview of AAVs based on of pathogenesis, clinical features, and diagnostic tools.
Eosinophilic Granulomatosis with Polyangiitis (EGPA)
It is an autoimmune reaction of unknown cause that produces blood vessels inflammation in patients with a pulmonary allergic history. The former name for EPGA is Churg-Strauss Syndrome (CSS), sometimes referred to as allergic granulomatosis. Among AAVs it is the rarest disease described by Churg and Strauss in 1951 characterized by MPO-ANCA presence in 30-38% of patients’ serum, and constant allergic reaction induced by IgE autoantibodies (Figure 1). This reaction has resulted in intensification of EGPA pathogenesis by marked infiltration of eosinophils into the affected vessels tissues. In addition, EGPA augmentation was promoted through increased production of IL-5 revealed by CD4+ cells and clonal expansion of CD8+ T cells [56]. Also, α-1-antitrypsin deficiency is observed in patient with EGPA [57].
EGPA Clinical Features and Management: A New Progress
It has been hypothesized that the ENT involvement in EGPA patients is an essential aspect. Therefore, nose and ear are the commonest sites where the clinical manifestations of EGPA are presented (Figure 1). Three stages of EPGA are recognized; (1) airway inflammation, (2) eosinophilia, and (3) cell death (vasculitis). Its prevalence is 2-22 per million persons and the annual incidence rate is 0.5-3.7 per million persons (58). Heart and cerebral lesions are considered as uncommon clinical manifestations of EPGA. However, attention should be paid whenever antiplatelet drugs recommended to such patients. Cardiac involvements in EPGA include myocarditis, heart failure, pericarditis, myocardial infarction, and constrictive pericarditis. In another topic, EGPA could be associated with inappropriate antidiuretic hormone syndrome [59]. EGPA categorized into two different clinical phenotypes according to the type/level of autoantibodies patient serum, ANCAnegative EGPA and anti-MPO-ANCA EGPA. The patients of the first subtype are more frequently to develop complications involving the heart whereas patients of the second subtype are more likely to develops classical vasculitis symptoms usually glomerulonephritis and alveolar hemorrhage [60].
The treatment of EPGA includes glucocorticoids, azathioprine, and cyclophosphamide. The management should include continuous clinical monitoring. In EGPA patients with moderate to severe allergic asthma, rituximab and omalizumab was considered effective and safe [11,61]. It has been reported that in EGPA and refractory asthma association the treatment by mepolizumab and omalizumab is a satisfactory option [62]. Meanwhile, heart transplantation in patients with refractory EGPA was recommended [63]. Samson and his co-workers recorded two important facts, the first is, the survival rate in EGPA patients is very good when treatment is managed by the baseline Five-Factor Score (FFS), and the second is, patients with p-ANCA and <3000/mm baseline eosinophilia are more exposed to relapses [64]. On the other hand, bullous phenotypes in relapsing EGPA especially the ANCAnegative form respond rapidly to systemic corticosteroids [65,66]. In progressive cases of EPGA, intravenous immunoglobulins (IVIGs) were effective in preventing the immobilization of neuropathy [65].
Microscopic Polyangiitis (MPA)
MPA is a member of small vessel vasculitis that often has a fatal outcome, and its histology shows little or no immune deposits. In MPA almost all types of blood vessels are clinically involved. The common organs involved in MPA are kidney and lung which usually followed by of nervous and musculoskeletal systems involvement. Skin, heart, eyes, and intestine are also involved. Although ANCA in MPA is mainly directed to MPO-ANCA (Figure 1), Immunoglobulin M (IgM) autoantibodies against PR3 were reported in MPA patients [67]. MPO-ANCA autoantibodies contribute to NETs formation, which might be involved in the pathophysiology of patients with MPA [68]. Also, exposure to silica was reported as a factor which could stimulate the producing mechanisms of MPA pathogenesis [69].
MPA Clinical Features and Management: A New Progress
Clinical manifestations of MPA are so variable including the pulmonary and renal involvement, and skin manifestations (Figure 1). Also, severe co-trimoxazole-induced hypoglycaemia, crescentic glomerulonephritis, acute cervical myelitis, and acute cholecystitis have been observed in patient with MPA [70-73]. Indeed, ruptured gastric artery aneurysm, hemorrhagic stroke, pyrexia of unknown origin, lymphoma, bilateral brachial plexopathy, intraventricular hemorrhage, and pneumomediastinum are recognizesd as the uncommon manifestations and complications of MPA [66,72- 75,77]. Meanwhile, patients with MPA are at risk for developing venous thromboembolic events (VTE) and eye involvement [78,79]. On the other hand, The Azathioprine hypersensitivity is presented as cardiogenic shock and weet’s syndrome in a patient with MPA [80]. In addition, MPA may be associated with fibrosing interstitial lung disease (ILD), primary biliary cirrhosis, Sjogren’s syndrome, and Hashimoto’s thyroiditis [81,82]. In Japanese patients, kidney survival was affected in relapsed MPA associated with infectious complication [83]. Indeed, phenotypes difference in patients with MPA from Europe and Japan has been explained [84].
Despite there are no standard treatment protocols for treatment of MPA patients, the effective communication between rheumatology, pulmonary, and nephrology physicians could improve disease management. Renal survival in MPA Chinese patients was promoted by glucocorticoids and mycophenolate mofetil (MMF) therapy [85]. However, MPA Japanese patients with renal involvement couldn’t get satisfactory results after treatment by cyclophosphamide and corticosteroid [86]. Other monoclonal antibodies, rituximab and Tocilizumab (TCZ) monotherapy could be a novel choice in some MPA patients [11,87]. Management protocols based on rituximab and MMF in children with MPA were achieved satisfactory outputs [88]. Meanwhile, MPA with cholecystitis elderly patient had been successfully treated using mizoribine [89]. As well, therapy directed at T cells might be an alternative treatment option for a rare MPA case with GIT involvement [90]. Unfortunately, disseminated mycetoma caused by Nocardia pseudobrasiliensis in MPA patient on long-term corticosteroid therapy was observed as a unique case in Korea [91]. Currently, gabexate by its antiinflammatory functions could be helpful in MPA management [92].
Concluding Remarks
The exact cause of AAVs is still not fully understood and we are yet to have complete remission. Patients with AAVs develop new severe symptoms. Therefore, continuous monitoring and evaluation remains critical to avert, control, or prevent unforeseen complications that could emanate from the disease. T cells subsets differentiation and their interleukins complements pathways activation, genetic basis, auto-reactivity, and environmental factors are the main contributors to AAVs pathogenesis. Recently, an advanced understanding of AAVs pathogenesis has led to the development and use of new therapeutic alternatives such as rituximab, tocilizumab, gabexate, omalizumab, mepolizumab, and mizoribine. However, the advancement in knowledge in AAVs suggested that effective collaboration between the different internal medicine sections specialists as well as radiologist and histopathologist is strongly recommended to optimize diagnosis and enhance the management of AAVs patients. Undoubtedly, future research focusing on the optimization of duration and frequency of maintenance therapy, and development of new therapeutic agents could help in the effective treatment and cure of the disease.
Author Contributions
All authors made significant contribution to the development of this manuscript.
Competing Interest
The authors declare that they have no competing interest. None of the authors has any financial or other interest influencing the output of this review.
Acknowledgement
The review was funded by Liaoning provincial program for top discipline of basic medical sciences; National Natural Science Foundation of China (81671606); Special Grant for Translational Medicine, Dalian Medical University (2015010); College Scientific Research Project of Education Department of Liaoning Province (LQ2017004); Liaoning Distinguished Professor (Liao taught (2018-2020)).
References
- Prendecki M, Pusey CD (2018) Recent advances in understanding of the pathogenesis of ANCA-associated vasculitis. F1000Research.
- Pagnoux C (2016) Updates in ANCA-associated vasculitis. Eur J Rheumatol 3(3): 122-133.
- Geetha D, Jin Q, Scott J, Zdenka Hruskova, Mohamad Hanouneh, et al. (2018) Comparisons of Guidelines and Recommendations on Managing Antineutrophil Cytoplasmic Antibody–Associated Vasculitis. Kidney International Reports 3(5): 1039-1049.
- Púechal X (2018) Targeted Immunotherapy Strategies in ANCA-Associated Vasculitis. Joint Bone Spine 86(3): 321-326.
- Kallenberg CG, Stegeman CA, Abdulahad WH, Heeringa P (2013) Pathogenesis of ANCA-associated vasculitis: new possibilities for intervention. Am J Kidney Dis 62(6): 1176-1187.
- Ramirez GA, Blasi M, Sciorati C, Rovere Querini P, Manfredi AA (2015) Plasma levels of M-CSF are increased in ANCA-associated vasculitides with active nephritis. Results Immunol 21(5): 33-36.
- Johansson ÅC, Ohlsson S, Pettersson Å, Bengtsson AA, Selga D, et al. (2016) Impaired phagocytosis and reactive oxygen species production in phagocytes is associated with systemic vasculitis. Arthritis Res Ther 18: 92.
- Bonatti F, Reina M, Neri TM, Martorana D (2014) Genetic Susceptibility to ANCA-Associated Vasculitis: State of the Art. Front Immunol 5: 577.
- Lally L, Spiera R (2015) Current therapies for ANCA-associated vasculitis. Annu Rev Med 66: 227-240.
- Schönermarck U, Gross WL, De Groot K (2014) Treatment of ANCA-associated vasculitis. Nat Rev Nephrol 10(1): 25-36.
- Daikeler T, Kistler AD, Martin PY, Vogt B, Huynh Do U (2015) The role of rituximab in the treatment of ANCA-associated vasculitides (AAV). Swiss Med Wkly 6(145): w14103.
- Luqmani RA (2014) State of the art in the treatment of systemic vasculitides. Front Immunol 13(5): 471.
- AL Azab M, Wei J, Ouyang X, Elkhide A, Walana W, et al. (2018) TL1A Mediates FLS Migration and Indian Hedgehog Signaling Pathway via TNFR2 in Rheumatoid Arthritis Patients. ECN 29(1): 27-35.
- Silva F, Cisternas M, Specks U (2012) TNF-α blocker therapy and solid malignancy risk in ANCA-associated vasculitis. Curr Rheumatol Rep 14(6): 501-508.
- Grayson PC, Cuthbertson D, Carette S, Hoffman GS, Khalidi NA, et al. (2013) New features of disease after diagnosis in 6 forms of systemic vasculitis. J Rheumatol 40(11): 1905-1912.
- Wegener F, Ubergeneralisierte, septische Gefaesserkrankungen (1936) Verhandlungen der Deutschen Gesellschaft fur Pathologie 29: 202-227.
- Wegener F (1939) Ubereineeigenartige Rhinogene Granulomatosemitbesondere Beteilgung des Arterien systems und der Nieren. Beitrage Pathologie Anatomie102: 36-51.
- Godman GC, Churg J (1954) Wegener’s granulomatosis: Pathology and review of the literature. AMA Archives of Pathology 58(6): 533-553.
- Falk RJ, Gross WL, Guillevin L (2011) Granulomatosis with polyangiitis (Wegener’s): An alternative name for Wegener’s granulomatosis. Annals of the Rheumatic Diseases 70: 704.
- Holle JU, Windmöller M, Lange C, Gross WL, Herlyn K, et al. (2013) Toll-like receptor TLR2 and TLR9 ligation triggers neutrophil activation in granulomatosis with polyangiitis. Rheumatology (Oxford) 52(7): 1183-1189.
- Bae S, Kim YG, Choi J, Hong J, Lee S, et al. (2012) Elevated interleukin-32 expression in granulomatosis with polyangiitis. Rheumatolog Fy (Oxford) 51(11): 1979-1988.
- Wilde B, Hua F, Dolff S, Jun C, Cai X, et al. (2012) Aberrant expression of the negative costimulator PD-1 on T cells in granulomatosis with polyangiitis. Rheumatology (Oxford) 51(7): 1188-1197.
- Surmiak MP, Hubalewska Mazgaj M, Wawrzycka Adamczyk K, Szczeklik W, Musiał J, et al. (2015) Circulating mitochondrial DNA in serum of patients with granulomatosis with polyangiitis. Clin ExpImmunol 181(1): 150-155.
- Surmiak M, Hubalewska Mazgaj M, Wawrzycka Adamczyk K, Szczeklik W, Musiał J, et al. (2016) Neutrophil-related and serum biomarkers in granulomatosis with polyangiitis support extracellular traps mechanism of the disease. Clin Exp Rheumatol 34(3 Suppl 97): S98-104.
- Inaty H, Arabelovic S (2013) α1-Antitrypsin deficiency in a patient diagnosed with granulomatosis with polyangiitis. BMJ Case Rep.
- Kopp SA, High WA, Green JJ (2012) Levamisole-induced Wegener's granulomatosis following contaminated cocaine abuse. Skinmed 10(4): 254-256.
- Halawani HM, Khalife M (2016) IMAGES IN CLINICAL MEDICINE. Gastrointestinal Complication of Granulomatosis with Polyangiitis. N Engl J Med 374(22): 2159.
- Jethava A, Ali S (2013) Ischemic stroke as a presenting feature of Wegener's granulomatosis. Conn Med 77(9): 533-555.
- Bohm M, Gonzalez Fernandez MI, Ozen S, Pistorio A, Dolezalova P, et al. (2014) Clinical features of childhood granulomatosis with polyangiitis (wegener'sgranulomatosis). Pediatr Rheumatol Online J 12: 18.
- Dinić MZ, Sekulović LK, Zolotarevski L, Zecević RD (2013) Fulminant Wegener's granulomatosis: a case report. Vojnosanit Pregl 70(9): 887-890.
- Varnier GC, Sebire N, Christov G, Eleftheriou D, Brogan PA (2016) Granulomatosis with polyangiitis mimicking infective endocarditis in an adolescent male. Clin Rheumatol 35(9): 2369-2372.
- Kumamoto M, Tomoda K, Furuya Y, Iwasa N, Ueno S, et al. (2016) Hypertrophic Pachymeningitis as a Delayed Complication of Granulomatosis with Polyangiitis. Intern Med 55(4): 413-417.
- Huang YH, Ro LS, Lyu RK, Chang HS, Wu YR, et al. (2015) Wegener's granulomatosis with nervous system involvement: a hospital-based study. Eur Neurol 73(3-4): 197-204.
- Harrison L, Mcnally J, Corbridge R (2016) Granulomatosis with polyangiitis affecting the skull base and manifesting as spontaneous skull base osteomyelitis. BMJ Case Rep.
- Greco A, Marinelli C, Fusconi M, GF Macri,2 A Gallo, et al. (2016) Clinic manifestations in granulomatosis with polyangiitis. International Journal of Immunopathology and Pharmacology 29(2): 151-159.
- Bîrluţiu V, Rezi EC, Bîrluţiu RM, Zaharie IS (2016) A rare association of chronic lymphocytic leukemia with c-ANCA-positive Wegener's granulomatosis: a case report. World J Surg Oncol 14: 145.
- Ghosh A, Banerjee A, Saha S, Pande A, Ghosh B (2012) Wegener's granulomatosis with dengue fever: an unusual association. Int J Rheum Dis 15(3): e47-e49.
- Jain P, Ruchin P, Suttie J (2016) Proximal pulmonary artery stenosis: a rare manifestation of granulomatosis with polyangiitis. Lancet 387(10035): 2349-2350.
- Ben Salah R, Frikha F, Snoussi M, Abderrahmen M, Hentati Y, et al. (2014) Limited form of Wegener's granulomatosis in a patient with Crohn's disease. A case report. Turk J Gastroenterol 25(1): 191-195.
- McGeoch L, Carette S, Cuthbertson D, Hoffman GS, Khalidi N, et al. (2015) Vasculitis Clinical Research Consortium. Cardiac Involvement in Granulomatosis with Polyangiitis. J Rheumatol 42(7): 1209-1212.
- Lacoste C, Mansencal N, Ben M’rad M, Goulon Goeau C, Cohen P, et al. (2011) Valvular involvement in ANCA-associated systemic vasculitis: a case report and literature review. BMC MusculoskeletDisord 12: 50.
- Ying CM, Yao DT, Ding HH, Yang CD (2014) Infective endocarditis with antineutrophil cytoplasmic antibody: report of 13 cases and literature review. PLoS One 9(2): e89777.
- Castellanos D, Travelli FC, Reyhan I, Votava Smith JK, Ramanathan A, et al. (2015) Acute Aortic and Mitral Valve Perforations Caused by Granulomatosis With Polyangiitis. Circulation 131(24): e527-e529.
- Langford CA, Monach PA, Specks U, Seo P, Cuthbertson D, et al. (2014) An open-label trial of abatacept (CTLA4-IG) in non-severe relapsing granulomatosis with polyangiitis (Wegener's). Ann Rheum Dis 73(7): 1376-1379.
- Ebrahimiadib N, Modjtahedi BS, Roohipoor R, Anesi SD, Foster CS (2016) Successful Treatment Strategies in Granulomatosis With Polyangiitis-Associated Peripheral Ulcerative Keratitis. Cornea 35(11): 1459-1465.
- Lu CW, Zhou DD, Wang J, Hao JL (2016) Surgical treatment of peripheral ulcerative keratitis and necrotizing scleritis in granulomatosis with polyangiitis. Saudi Med J 37(2): 205-207.
- Sikorska D, Tykarski A, Radziemski A, Mojs E, Samborski W (2015) Atypical location of granulomatosis with polyangiitis (Wegener's) with heart involvement--effectiveness of treatment with rituximab. Kardiol Pol 73(12): 1338.
- Besada E (2016) Low immunoglobulin levels increase the risk of severe hypogammaglobulinemia in granulomatosis with polyangiitis patients receiving rituximab. BMC Musculoskelet Disord 17: 6.
- Malik M, Ismail M, Pattanaik D (2015) Granulomatosis With Polyangiitis Presenting as Gastric Ulcer: An Unusual Initial Manifestation Successfully Treated With Rituximab. Am J Med Sci 350(4): 338-339.
- Sandhu RK, Adams T, Sibley C, Suhler EB, Kim DH (2016) Granulomatosis With Polyangiitis (Gpa) Presenting With Frosted Branch Angiitis. Retin Cases Brief Rep 10(3): 249-251.
- Gastman B, HashemAM, Djohan R, Bernard S, Hendrickson M, et al. (2016) Malignant Pyoderma Associated with Granulomatosis with Polyangiitis (Wegener Granulomatosis) as a Unique Indication for Facial Vascularized Composite Allotransplantation: Part I. Plast Reconstr Surg 137(6): 1007e-1015e.
- Horta Baas G, Hernández Cabrera MF, Catana R, Pérez Cristóbal M, Barile Fabris LA (2016) Subglottic stenosis in granulomatosis with polyangiitis (Wegener's granulomatosis): Report of 4 cases. Reumatol Clin 12(5): 267-273.
- Lee IH, Kang GW, Kim KC (2016) Hypersensitivity pneumonitis associated with azathioprine therapy in a patient with granulomatosis with polyangiitis. Rheumatol Int 36(7): 1027-1032.
- Ayoub I, Almaani S, Alvarado A, Parikh SV, Rovin BH (2016) Quiz Page February 2016: Acute Kidney Injury in a Patient With Granulomatosis With Polyangiitis Receiving Maintenance Immunosuppressive Therapy. Am J Kidney Dis 67(2): A20-A30.
- Fukui S, Iwamoto N, Tsuji S, Umeda M, Nishino A, et al. (2015) Diffuse alveolar hemorrhage emerging one week after starting high-dose corticosteroid therapy for granulomatosis with polyangiitis (GPA) with systemic lupus erythematosus (SLE). Intern Med 54(20): 2681-2686.
- Boita M, Guida G, Circosta P, Elia AR, Stella S, et al. (2014) The molecular and functional characterization of clonally expanded CD8+ TCR BV T cells in eosinophilic granulomatosis with polyangiitis (EGPA). Clin Immunol 152(1-2): 152-163.
- Moxey JM, Low EV, Turner AM (2016) Rare case of eosinophilic granulomatosis with polyangiitis in two patients with α-1-antitrypsin deficiency (PiSZ). BMJ Case Rep.
- Mahr A, Moosig F, Neumann T, Szczeklik W, Taillé C, et al. (2014) Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): evolutions in classification, etiopathogenesis, assessment and management. Curr Opin Rheumatol 26(1):16-23.
- Tokushige S, Kodama K, Hideyama T, Kumekawa H, Shimizu J, et al. (2016) Syndrome of Inappropriate Antidiuretic Hormone Associated with Eosinophilic Granulomatosis with Polyangiitis. Intern Med 55(9): 1199-1202.
- Mouthon L, Dunogue B, Guillevin L (2014) Diagnosis and classification of eosinophilic granulomatosis with polyangiitis (formerly named Churg-Strauss syndrome). J Autoimmun 48-49: 99-103.
- Detoraki A, Di Capua L, Varricchi G, Genovese A, Marone G, et al. (2016) Omalizumab in patients with eosinophilic granulomatosis with polyangiitis: a 36-month follow-up study. J Asthma 53(2): 201-216.
- Lutalo PM, D'Cruz DP (2015) Biological drugs in ANCA-associated vasculitis. Int Immunopharmacol 27(2): 209-212.
- Groh M, Masciocco G, Kirchner E, Kristen A, Pellegrini C, et al. (2014) Heart transplantation in patients with eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome). J Heart Lung Transplant 33(8): 842-850.
- Samson M, Puéchal X, Devilliers H, Ribi C, Cohen P, et al. (2013) Long-term outcomes of 118 patients with eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome) enrolled in two prospective trials. J Autoimmun 43: 60-69.
- Sherman S, Gal N, Didkovsky E, Molad Y, Pavlovsky L, et al. (2017) Eosinophilic Granulomatosis with Polyangiitis (Churg-Strauss) Relapsing as Bullous Eruption. Acta Derm Venereol 97(3): 406-407.
- Matsumoto T, Otsuka K, Kawamoto M, Nagata K, Tachikawa R, et al. (2013) Efficacy of early intravenous immunoglobulin for eosinophilic granulomatosis with polyangiitis with drastically progressive neuropathy: a synopsis of two cases. Intern Med 52(8): 913-917.
- Clain JM, Hummel AM, Stone JH, Fervenza FC, Hoffman GS, et al. (2017) Immunoglobulin (Ig)M antibodies to proteinase 3 in granulomatosis with polyangiitis and microscopic polyangiitis. Clin Exp Immunol 188(1): 174-181.
- Yoshida M, Yamada M, Sudo Y, Kojima T, Tomiyasu T, et al. (2016) Myeloperoxidase anti-neutrophil cytoplasmic antibody affinity is associated with the formation of neutrophil extracellular traps in the kidney and vasculitis activity in myeloperoxidase anti-neutrophil cytoplasmic antibody-associated microscopic polyangiitis. Nephrology 21(7):624-629.
- Vega Miranda J, Pinto Peñaranda LF, Márquez Hernández JD, Velásquez Franco CJ (2014) Microscopic polyangiitis secondary to silica exposure. Reumatol Clin 10(3): 180-182.
- Conley TE, Mohiuddin A, Naz N (2017) Severe co-trimoxazole-induced hypoglycaemiain a patient with microscopic polyangiitis. BMJ Case Rep.
- Cho AY, Kim BG, Kim SS, Lee SH, Shin HS, et al. (2016) Microscopic polyangiitis with crescentic glomerulonephritis initially presenting as acute pancreatitis. Korean J Intern Med 31(2): 403-405.
- ToudouDaouda M, Obenda NS, Camara D, El Midaoui A, Belahsen MF, et al. (2016) Acute cervical myelitis and microscopic polyangiitis. Rev Neurol 172(4-5): 325-327.
- Ikura Y, Kadota T, Watanabe S, Arimoto A, Nishioka E (2014) Ruptured gastric artery aneurysm: an uncommon manifestation of microscopic polyangiitis. World J Gastroenterol 20(35): 12668-12672.
- Ziaj S, Mitchell C, Roufosse C, Dubrey SW (2014) Occult microscopic polyangiitis presenting as pyrexia of unknown origin. Br J Hosp Med 75(3): 172-173.
- Huang YH (2015) Microscopic Polyangiitis Manifesting as Lymphoma. Ocul Immunol Inflamm 23(3): 256-258.
- NaseriAlavi SA, Meshkini M, Pourlak T, Khabbazi A (2016) Microscopic polyangiitis complicated with bilateral brachial plexopathy: a case report and review of the literature. Rheumatol Int 36(7): 997-1001.
- Isoda K, Takeuchi T, Ishida T, Makino S, Hanafusa T (2014) Pneumomediastinum in a patient with microscopic polyangiitis preceded by interstitial pneumonia. Intern Med 53(8): 891-893.
- Kapoor S (2013) Ophthalmic complications of microscopic polyangiitis: an often overlooked disease entity. J Stroke Cerebrovasc Dis 22(6): 894-895.
- Kechaou I, Cherif E, Boukhris I, Azzabi S, Kaouech Z, et al. (2015) Microscopic polyangiitis: A little-known new risk factor of venous thrombosis. J Mal Vasc 40(6): 406-407.
- Turow A, Yong TY, Fok JS, Li JY (2012) Azathioprine hypersensitivity presenting as cardiogenic shock and Sweet's syndrome in a patient with microscopic polyangiitis. Intern Med 51(14): 1889-1892.
- Schirmer JH, Wright MN, Vonthein R, Herrmann K, Nölle B, et al. (2016) Clinical presentation and long-term outcome of 144 patients with microscopic polyangiitis in a monocentric German cohort. Rheumatology 55(1): 71-79.
- Ghorbel IB, Feki NB, Salem TB, Hamzaoui A, Khanfir M, et al. (2015) Microscopic polyangiitis associated with primary biliary cirrhosis, Sjogren's syndrome and Hashimoto's thyroiditis. Saudi J Kidney Dis Transpl 26(2): 359-362.
- Kitagawa K, Furuichi K, Sagara A, Shinozaki Y, Kitajima S, et al. (2016) Kanazawa Study Group for RenalDiseases and Hypertension. Risk factors associated with relapse or infectious complications in Japanese patients with microscopic polyangiitis. Clin Exp Nephrol 20(5): 703-711.
- Furuta S, Chaudhry AN, Hamano Y, Fujimoto S, Nagafuchi H, et al. (2014) Comparison of phenotype and outcome in microscopic polyangiitis between Europe and Japan. J Rheumatol 41(2): 325-333.
- Chen Y, Gao E, Yang L, Liu X, Li K, et al. (2016) Long-term outcome of mycophenolate mofetil treatment for patients with microscopic polyangiitis: an observational study in Chinese patients. Rheumatol Int 36(7): 967-974.
- Iwabuchi M, Nakaya I, Tsuchiya Y, Shibagaki Y, Yamaguchi T, et al. (2016) Effects of cyclophosphamide on the prognosis of Japanese patients with renal vasculitis associated with anti-neutrophil cytoplasmic antibody-positive microscopic polyangiitis. Clin Exp Nephrol 20(5): 712-719.
- Sakai R, Kondo T, Kikuchi J, Shibata A, Chino K, et al. (2016) Corticosteroid-free treatment of tocilizumab monotherapy for microscopic polyangiitis: a single-arm, single-center, clinical trial. Mod Rheumatol 26(6): 900-907.
- Basu B, Mahapatra TK, Mondal N (2015) Favourable renal survival in paediatric microscopic polyangiitis: efficacy of a novel treatment algorithm. Nephrol Dial Transplant 30(1): i113-i118.
- Ichinose K, Iwanaga N, Okada A, Tamai M, Yamasaki S, et al. (2014) A case of microscopic polyangiitis in an elderly patient presenting predominantly with cholecystitis successfully treated with mizoribine. Mod Rheumatol 24(6): 1011-1014.
- Latus J, Koetter I, Fritz P, Kimmel M, Biegger D, et al. (2014) Gastrointestinal involvement in granulomatosis with polyangiitis and microscopic polyangiitis: histological features and outcome. Int J Rheum Dis 17(4): 412-419.
- Seol CA, Sung H, Kim DH, Ji M, Chong YP, et al. (2013) The first Korean case of disseminated mycetoma caused by Nocardia pseudobrasiliensis in a patient on long-term corticosteroid therapy for the treatment of microscopic polyangiitis. Ann Lab Med 33(3): 203-207.
- Gigante A, Gasperini ML, Barbano B, Liberatori M, Sardo L, et al. (2013) Gabexate mesylate as treatment in the course of ANCA-negative microscopic polyangiitis. Ren Fail 35(5): 721-724.
