info@biomedres.us
+1 (502) 904-2126
One Westbrook Corporate Center, Suite 300, Westchester, IL 60154, USA
Site Map
Received: September 29, 2018; Published: October 09, 2018
*Corresponding author: Moniruzzaman, Faculty of Engineering, Department of Applied Chemistry and Biochemical Engineering, Shizuoka University, 3-5-1, Johoku, Hamamatsu, 432-8561, Japan
DOI: 10.26717/BJSTR.2018.09.001852
Most of the nonsteroidal anti-inflammation drugs (NSAID) have some demerits depending on type and nature of physical conditions and limit of doses. Herein, we report the optimization of Naproxen and its degradants employing density functional theory (DFT) with B3LYP/6-31g+(d,p) level theory to elucidate their thermal and molecular orbital properties. Molecular docking and nonbonding interactions have been performed against prostaglandin synthase protein (5F19) to search binding affinity and interactions of all compounds with the respective protein. Pharmacokinetic properties also calculated to search their absorption, metabolism, and carcinogenicity.
Abbreviations: COX: Cyclooxygenase; DFT: Density Functional Theory; NSAID: Nonsteroidal Anti-Inflammation Drugs; PGH2: Prostaglandin H2; HOMO: Highest Occupied Molecular Orbital; LUMO: Lowest Unoccupied Molecular Orbital; QM: Quantum Mechanical; LYP: Lee, Yang and Parr's; PDB: Protein Data Bank; SDF; Structure Data File; SMILES: Simplified Molecular-Input Line-Entry System; hERG: Human Ether-A-Go-Go-Related Gene; BBB: Blood Brain Barrier
Figure 1: Chemical structure of Naproxen (Nap) and its degra-dants.

Naproxen is a naphthalene nucleus bearing nonsteroidal an- ti-pyratic and anti-inflammatory drug, that plays key role against cyclooxygenase (COX) leading to suppress prostaglandins accumulation caused by various diseases [1,2]. It has undesirable side effects upon routine medication due to free form of terminal acid group. A bunch of modification focused on terminal acid group protection or prodrug derivatization in order to minimize the secondary effect [1-3]. Several degradative studies suggested to investigate the nature of degradant in the different chemical and physical environment [4,5]. In this study, we considered all the four impurities including degradants such as D1, D2, D3, and D4 with that parent Naproxen. Here D2 and D4 degradative products were obtained during acid hydrolysis (1N HCl at 60°C for 2hrs) and base hydrolysis (1N NaOH at 60°C for 6hrs) where as D1 and D3 were considered as metabolite and process related by products [4]. Previously, computational studies of Naproxen and some of its modified derivatives also reported [6,7] (Figure 1).
Figure 2: Frontier molecular orbital (HOMO-LUMO) and related transition energy of Naproxen and D1.

In this investigation, we report the optimization and prostaglandin H2 (PGH2) inhibition pathway of Naproxen and its degradants utilizing molecular docking, nonbonding interactions, and pharmacokinetic calculations. Enthalpy, free energy, dipole moment, HOMO (highest occupied molecular orbital), LUMO (lowest unoccupied molecular orbital), hardness, softness, chemical potential has and chemical potential have been studied for every molecule. Molecular docking and nonbonding interactions calculation are performed to understand the binding affinity and binding mode(s) of all structures with the receptor protein (5F19). Some of the compounds show improved thermal, molecular orbital and binding properties (Figure 2).
Quantum mechanical (QM) methods keep an important role for the calculation of thermal and molecular orbital properties [8]. In this investigation, QM calculation was implemented by using density functional theory (DFT) employing Becke's (B) [9] exchange functional combining Lee, Yang and Parr's (LYP) correlation functional [10] in Gaussian 09 program package for all compounds [11]. People's 6-31g + (d,p) basis set was used to optimize the drugs and other calculations [12]. Initial geometry of Naproxen was taken from online chemical information resource named ChemSpider and further modified in Gaussian 09 software[13]. For every molecule’s internal electronic energy, enthalpy, Gibb's free energy and dipole moment were calculated. Frontier molecular orbital calculation was performed by using same level of theory. Hardness (ƞ) and softness of all drugs were also calculated from the energies of frontier HOMOs and LUMOs considering Parr and Pearson interpretation [14,15] of DFT and Koopmans theorem [16] on the correlation of ionization potential (I) and electron affinities (E) with HOMO and LUMO energy (Ɛ). The following equations are used for the calculation of hardness (ƞ) and softness (S):
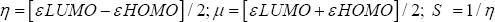
Three-dimensional crystal structure of aspirin acetylated human cyclooxygenase-2 (PDB ID: 5F19) was retrieved in PDB format from online protein data bank (PDB) [17]. All hetero atoms and water molecules were eliminated using PyMol (version 1.3) software packages [18]. Energy minimization of the protein implemented by Swiss-Pdb viewer software (version 4.1.0) [19]. Than optimized drugs were subjected for molecular docking study against human prostaglandin synthase protein (5F19). In computer aided drug design, binding affinity and mode(s) of ligand with target protein can predict by molecular docking simulation [20,21]. Finally, molecular docking simulation was performed by PyRx software (version 0.8) [22] considering the protein as macromolecule and the drug as ligand. In this analysis, rigid docking was performed where, all rotatable bonds were converted into non-rotatable with the center grid box size 64.8642, 73.2984, and 57.9414 A along x, y and z directions respectively. After docking, both the protein and ligand structures were saved in. pdbqt format required by Accelrys Discovery Studio (version 4.1) to analyze and visualize the docking result and search the interactions between ligands and amino acid residues of receptor protein [23].
Absorption, metabolism and carcinogenicity of Naproxen and its derivatives were predicted by utilizing AdmetSAR online database [24]. SDF (Structure Data File) and SMILES (simplified molecular-input line-entry system) strings were used throughout the generation process (Table 4).
Figure 3: (A) Docked conformation of all structures at inhibition bounding site of 5F19 (B). Superimposed view of all compounds after rigid docking.

The spontaneity of a chemical reaction and the stability of the reaction product can be predicted from thermodynamic properties such as enthalpy, Gibb's free energy [25]. Free energy is a pivotal criterion to represent the interactions of binding partners where both the sign and magnitude are important to express the likelihood of bimolecular events occurring. Greater negative values indicate improved thermodynamic properties. In this study, it is found that the values are negative (Table 1) meaning the binding will occur spontaneously without any extra energy expenditure [26]. Free energy of Naproxen is -767.4245 Hartree where D3 shows the highest values (-882.7084 Hartree) which suggesting that the molecules are energetically and configurationally more preferable. The dipole moment of Naproxen is 3.1812 sDebye where D3 shows the highest dipole moment (4.3669 Debye) (Figure 4). Elevated level of dipole moment enhances the hydrogen bond formation, nonbonding interaction, binding affinity and polar nature of a molecule [27].
Table 1: Molecular formula, molecular weight, enthalpy, free energy (in Hartree), and dipole moment (Debye) of all compounds.

Figure 4: Nonbonding interactions of Naproxen (Nap) and D3 with the amino acid residues of 5F19 generated by Discovery Studio.

Chemical hardness (ƞ) and softness (S) of a molecule can determine from the HOMO (highest occupied molecular orbital) - LUMO (lowest unoccupied molecular orbital) gap [28]. Large HOMO-LUMO gap related to high kinetic stability and low chemical reactivity and small HOMO-LUMO gap is important for low chemical stability, because addition of electrons to a high-lying LUMO and/ or removal of electrons from a low-lying HOMO is energetically favorable in any potential reaction [29]. In this study, Naproxen has the HOMO-LUMO gap 4.4665 eV where D3 shows the lowest energy gap (2.0028 eV) and the lowest chemical potential (-3.7252 eV) with the highest chemical softness (0.9986 eV) values which may contribute the higher chemical reactivity than others (Table 2). Table 2: HOMO, LUMO, gap, hardness, and softness of all compounds.
Table 2: HOMO, LUMO, gap, hardness, and softness of all compounds.

Naproxen showed a binding energy of -9.3 kcal/mol whereas increasing order of binding energy followed the sequence such as D2 Table 3: Binding affinity and nonbonding interactions of all compounds. Table 4: Selected pharmacokinetic parameters of Naproxen and its degradants. From AdmetSAR calculation, it is found that all the drugs show positive response for blood brain barrier (BBB) criteria, predicting that drugs can pass through the BBB. All the drugs show III category acute oral toxicity so, it can be predicted that they are relatively harmless for oral administration. AdmetSAR calculation predicts all drugs are non-carcinogenic. So, the drugs are expected to be safe for topical use. All drugs are P-glycoprotein non-inhibitor. P-glycoprotein inhibition can block the absorption, permeability and retention of the drugs [30]. However, all the molecules show non-inhibitory property for human ether-a-go-go-related gene (hERG). Inhibition of hERG can lead to long QT syndrome [31], so more study of this aspect is necessary. In this investigation, the inherent stability and biochemical interaction of Naproxen and its degradants have been studied. From, DFT calculation all the compounds are thermally stable and some of them show better chemical reactivity than Naproxen. D4 show greater dipole moment with smaller HOMO-LUMO gap. Apart from that, D3-5F19 complex shows better binding affinity with significant interactions than others. Pharmacokinetic results predict all the degradants are non-carcinogenic and relatively safe for oral administration. Considering above discussion, this study may be helpful to understand the physiochemical and biological properties of Naproxen and its degradants. Authors are grateful to Dr. Mohammad A. Halim, COE, The Red Green Research Centre, Dhaka, Bangladesh for his intellectual support during optimization.


Pharmacokinetic Investigation
Conclusion
Acknowledgement
Acknowledgement

























