info@biomedres.us
+1 (502) 904-2126
One Westbrook Corporate Center, Suite 300, Westchester, IL 60154, USA
Site Map
Received: January 20, 2018; Published: January 30, 2018
Corresponding author: Sunil Ashrani, M. Pharma Research Scholar, Bhupal Nobles' University, Sewashram Choraya, Udaipur, Rajastan, India
DOI: 10.26717/BJSTR.2018.02.000704
ICH Q8 guideline defines Quality by Design as "a systematic approach to development that begins with predefined objectives and emphasizes product and process understanding and process control, based on sound science and quality risk management. The QbD based approaches provides a high degree of assurance that pharmaceutical manufacturing process is adjustable within a design space and hence the process is robust and managed with a control strategy developed using modern statistical process control methods. It enables a lifecycle approach to validation/continuous process verification. The building of Quality into the manufacturing products at its design stage is very important to reduce expenditure, time & energy. Also the market recall of products due to batch failure is reduced to minimum with the use of QbD. PAT is also related to QbD. It refers to Process analytical technology. PAT has been defined as "A system for designing, analyzing, and controlling manufacturing through measurements, during processing of critical quality and performance attributes of raw and in-process materials and processes, with the goal of ensuring final product quality”. The present paper deals on these two terms QbD and PAT.
Keywords: Quality by Design; PAT; Design Stage; ICH Q8
On looking to the scenario in pharmaceutical industries we find that since past few decades, pharmaceutical companies had been spending very large amount of resources to built quality, achieve regulatory compliance, and produce drugs as cost effectively as possible. For this they employ advance processes and technologies. But such efforts have not been so effective. It is due to the of lack of comprehensive, rationale based understanding of these processes, associated critical variables and strategies to control these variables, which is important in assuring quality of the product. The companies show less interest in identifying the root cause of manufacturing failures. Furthermore, no rationale-based approach is followed to predict the effects of scale-up on the final product [1]. But with changing time and increasing complexity of regulatory requirements in the approval of a drug to come in market, companies are focusing on methods through which they can prepare a design of the product and do researches related to complexities that may have to be faced in future and thus find out the solutions prior to manufacturing. This study is done through various tools like QbD, PAT etc. The main reason behind is only one and that is to reduce the cost of manufacturing defects (Table 1).
Table 1: Differences between current approach and QbD approach [5].

The improvement in increasing order without any strategy may not always give positive fruitful results. It would only have little effect on overall process performance or quality. Thus to assure the quality of the product, a more realistic approach is provided by QbD. QbD (Quality by Design) is defined in the ICH Q8 guideline as "a systematic approach to development that begins with predefined objectives and emphasizes product and process understanding and process control, based on sound science and quality risk management" [2,3]. The ideal QbD-based pharmaceutical development effort involves a systematic method relating mechanistic understanding of input material attributes and process parameters to drug product critical quality attributes. Such a development efforts are possible through the use of multivariate experiments that involve modern process controls and enables process understanding (Figures 1 & 2). QbD identifies parameters that are critical to quality from the perspective of patients, translates them into the attributes that the drug product must possess, and also establishes how the critical process parameters can be changed to consistently produce a drug product having the desired characteristics [4,5].
Figure 1: Illustration of the different steps in development of a pharmaceutical product.
Pharmaceutical Quality by Design [3].

Figure 2: Elements of Quality by Design [8].

For industry
a) It helps in better understanding of the process.
b) It reduces batch failure.
c) It ensures better design of products with fewer problems in manufacturing.
d) It allows for continuous improvement in products & manufacturing process.
a) It enhances scientific base for analysis.
b) It provides better consistency.
c) It provides more flexibility in decision making.
d) It ensures decisions are made on scientific base & not on observed information.
a. It is started with a target product profile that illustrates the use, safety and efficacy of the product
b. Then, introduces a target product quality profile that the formulators and process engineers use as a quantitative surrogate for aspects of clinical safety and efficacy of the product during development.
c. The collection of relevant prior knowledge about the drug substance, potential excipients and process operations into a knowledge space is also done.
d. Application of risk assessment tools to prioritize knowledge gaps for further investigation is necessary.
e. Formulation of a design to find the critical material (quality) attributes of the final product that is necessary to be controlled to meet the target product quality profile is then done.
f. Also formulate the design of manufacturing process to produce a final product having the required critical materials attributes.
g. Find out the critical process parameters and raw material attributes that should be controlled to achieve these critical material attributes of the final product.
h. Risk assessment must be used to prioritize process parameters and material attributes for experimental verification.
i. Combination of prior knowledge with experiments is important to establish a design space or other representation of process understanding.
j. Making of a control strategy for the entire process that must include raw material controls, process controls and monitors, design spaces around individual or multiple unit operations, and/or final product tests.
k. The control strategy must encompass expected changes in scale and can be guided by a risk assessment.
l. Monitoring and update of the process to assure consistent quality continually.
TPQP has been defined as a "prospective and dynamic summary of the quality characteristics of a drug product that ideally will be achieved to ensure that the desired quality, and thus the safety and efficacy, of a drug product is realized". It covers dosage form and route of administration, dosage form strength(s), therapeutic moiety release or delivery and pharmacokinetic characteristics (e.g., dissolution and aerodynamic performance) appropriate to the drug product dosage form being developed and drug product- quality criteria (e.g. sterility and purity) appropriate for the intended marketed product [8].
TPP forms the basis for product design in the following way [9].
a. Dosage form
b. Route of administration
c. Strength, maximum and minimum
d. Release/delivery of the drug
e. Pharmacological characteristic
f. Drug product quality criteria
g. Pharmaceutical elegance
A CQA has been defined as "a physical, chemical, biological or microbiological property or characteristics that should be within an appropriate limit, range, or distribution to ensure the desired product quality." According to ICHQ9 the Identification of CQAs is done through risk assessment. Critical Quality Attributes are associated with the drug substance, excipients, intermediates and drug product. Critical Quality attributes covers the properties that impart the desired quality, safety, and efficacy. In context of biotechnological products, CQAs are typically those aspects which affect product purity and stability. Drug product CQAs can be identified from the Target product profile. The use of strong risk assessment methods for identification of CQAs is new to the QbD standard [6,10].
Critical process parameters (CPPs) are defined as "parameters whose variability have an impact on a CQA and therefore should be monitored or controlled to ensure the process produces the desired quality" Process robustness is the ability of a process to demonstrate acceptable quality and performance and tolerate variability in inputs at the same time. To demonstrate the reproducibility and consistency of a process, process capability should be studied (Figures 3 & 4). Process capability is a statistical measure of the inherent process variability for a given characteristics. The most widely accepted formula for process capability is six-sigma [11]. Process capability index is the value of the tolerance specified for a particular characteristic divided by the process capability, which is defined as follows:
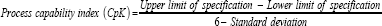
Figure 3: Design Space [15].

Figure 4: Flow Chart of Quality by Design [18].

If the CpK is significantly greater than one, the process is defined capable. But if the process capability is low, there are five step procedures to progressively reduce the variability of the process. These five steps are:
I. Define: The intended improvement should be clearly stated
II. Measure: The critical product performance attributes should be measured to see if they are out of specification and used to the sigma level of the process.
III. Analyze: When the sigma level is below the target, steps should be taken to increase it, starting by identifying the most significant causes of the excessive variability.
IV. Improve: The process should be redesigned and/ or process controls should be incorporated to eliminate or attenuate the significant toot causes of variance.
V. Control: The improved manufacturing process should be evaluated and maintained.
Risk assessment is the linkages between material attributes & process parameters. It is performed during the lifecycle of the product to identify the critical material attributes & critical process parameters. A material attributes can be an excipients raw material, drug substances, reagents, solvents, packaging & labelling materials. A material attributes can be quantified & typically fixed but sometimes can be changed during further processing [12].
E.g. Impurity profile, porosity, specific volume, sterility.
Design of experiments (DOE) is a systematic method to determine the relationship between factors affecting a process and the output of that process. In other words, it is used to find cause- and-effect relationships. This information is needed to manage process inputs in order to optimize the output. An understanding of DOE first requires knowledge of some statistical tools and experimentation concepts. Although a DOE can be analyzed in many software programs, it is important for practitioners to understand basic DOE concepts for proper application. [vii] For example parameters that affect the coating process are given in (Figure 5) [13,14]. Critical parameters are considered as independent variables in DOE.
Figure 5: Interrelationships between PAT and QbD [19].

ICH Q8 (R2) defines Design space as, the multidimensional combination and interaction of input variables (e.g. material attributes) and process parameters that have been demonstrated for provide assurance of quality. It will Working within the Design space is not be considered as a change, Movement out of the Design space it is considered to be a change and would normally initiate a regulatory post-approval change process. Design space is proposed by the applicant and is subject to regulatory assessment and approval. Thus Design space is potentially scale and equipment dependent, the Design space determined at the laboratory scale may not be relevant to the process at the commercial scale [14,15].
The concept originates from the desire of the regulators to shift control of product quality towards a science-based approach that explicitly attempts to reduce the risk to patients by controlling the manufacturing based on understanding of the process [16,17].
From a PAT standpoint, a process is considered well understood when:
a. All critical sources of variability are identified and explained;
b. Variability is managed by the process; and
c. Product quality attributes can be accurately and reliably predicted.
PAT has been defined as 'A system for designing, analyzing, and controlling manufacturing through measurements, during processing of critical quality and performance attributes of raw and in-process materials and processes, with the goal of ensuring final product quality”. The goal of PAT is to "enhance understanding and control the manufacturing process, which is consistent with our current drug quality system: quality cannot be tested into products; it should be built-in or should be by design.”
Quality has become an important issue in today's era. Everyone talks on quality but building quality is not mere task. It needs a long process for fulfilment of the required quality attributes in the material. When the term Quality is defined in context of Pharma, it becomes a legal issue. There are numerous guidelines and governing bodies who imply "should be and should not be conditions”. Quality by Design and Process analytical technology has been emerging concepts for developing the required quality attributes in the product at its design stage which saves time, energy and also reduces market recall of the products.

























