info@biomedres.us
+1 (502) 904-2126
One Westbrook Corporate Center, Suite 300, Westchester, IL 60154, USA
Site Map
Received: January 17, 2018; Published: January 23, 2018
*Corresponding author: Rua dos Jasmins, 326. Jardim Laguna, Linhares – Espírito Santo, Brazil
DOI: 10.26717/BJSTR.2018.02.000684
Diabetes insipidus is mainly characterized by polyuria, urinary volume over 3 L/day or 40mL/kg/day in adults, leading to subsequent polydipsia; these features are also present in most cases of diabetes mellitus. We report the case of a 56-year-old man initially misdiagnosed with and treated for diabetes mellitus; he was eventually diagnosed with central diabetes insipidus following further laboratory tests. We have also conducted a thorough review of the literature relating to the physiology, diagnosis, and treatment of diabetes insipidus. The patient, who complained of polyuria, polydipsia and weight loss, and had fasting glucose levels of 108 mg/dL, was already using metformin to treat type 2 diabetes. He then developed hypoglycemic symptoms, and pre- and postprandial capillary glycaemia between 70 and 120 mg/dL, as a result of which metformin was suspended. Nevertheless, polyuria and polydipsia persisted. Based on a plasma osmolality of 305,5 mOsm/kg, urinary density of 1005 g/mL and low arginine vasopressin levels, a diagnosis of central diabetes insipidus was made and treatment with desmopressin was initiated. Because the symptoms of centraldiabetes insipidus and uncontrolled type 2 diabetes mellitus overlap, it is important to consider clinical presentations carefully in order to make a differential diagnosis.
Keywords: Central Diabetes Insipidus; Osmolality; Desmopressin; Polyuria; Polydipsia
Abbreviations: DI: Diabetes insipidus; AVP : A Vasopressin; DM: Diabetes Mellitus; CDI: Central Diabetes Insipidus; PP: Primary Polydipsia
Diabetes insipidus (DI) is characterized by profuse diuresis, usually greater than 3L/24h of urine, with osmolality below 300mOsm/kg [1]. It has two main forms: Central (also known asneurogenic) DI is characterized by low levels of antidiuretic hormone, also known as vasopressin (AVP)[2]; nephrogenic DI is characterized by difficulties concentrating urine due to resistance to AVP action in the kidneys. AVP is the main determiner of free water clearance in the body. It acts mainly in the kidneys where it alters water permeability in the cortical tubules and the medulla. Water is reabsorbed by osmosisdue to hypertonicity within the medullary interstitium, and then taken back into systemic circulation [3]. Two other forms of DI are gestational DI and primary polydipsia (dipsogenic DI). Neither of these results from a defect in the neurohypophysis or the kidneys, but both participate in differential diagnoses of diabetes. The predominant manifestations of DI are polyuria, polydipsia, and nocturia, which also indicate diabetes mellitus (DM), another important differential diagnosis.
Laboratory tests, particularly 24-hour urine collection to determine the volume of urine and concentration of serum electrolytes, urine and plasma osmolality, and glucose and AVP levels, must be carefully analyzed,as small differences are crucial to a correct diagnosis. We report a case of central diabetes insipidus (CDI), accompanied by a literary review. CDI has many possible causes. According to the literature, the main causes of CDI and their frequencies are: idiopathic-30%; malign or benign tumors of the brain or neurohypophysis-25%; cranial surgery0-20%; and cranial trauma-16% [4]. As this disease is often undiagnosed, we will stress the importance of noting discrete abnormalities in laboratory results to make a differential, early diagnosis so that appropriate therapy can be started to improve the patient’s quality of life.
The patient, a 56-year-old Caucasian male, was referred to a tertiary endocrinology clinic by a Family Health Program physician. He had a history of mild systemic arterial hypertension and DM type2 (DM2) diagnosed five years previously. He spent two years receiving treatment for DM2, and experienced frequent episodes of hypoglycemia, in addition to benign prostatic hyperplasia. The patient had experienced frequent episodes of hypoglycemia, as a result of which he had had two emergency hospital visits at which he reportedtaking metformin (2g/day in two doses), enalapril (10mg/ day), and doxazosin (2mg/day). Capillary glycemia varied between 28 and 236mg/dL. His main concerns were hypoglycemia and intense thirst. At our initial consultation, on 25 September, 2015, he was currently using metformin 500mg/day; he weighed 87.1kg, his height was 1.72 m, and his body mass index (BMI) was 29.4kg/m2. Laboratory tests were requested, pre- and postprandial glycemia were monitored, and medication was maintained. Two months later (May 5, 2015), the patient returned to clinic asymptomatic. Glycaemia varied between 81 and 166 mg/dL. Furthertest results are shown in (Tables 1 & 2).
Table 1: Results of Blood Tests.
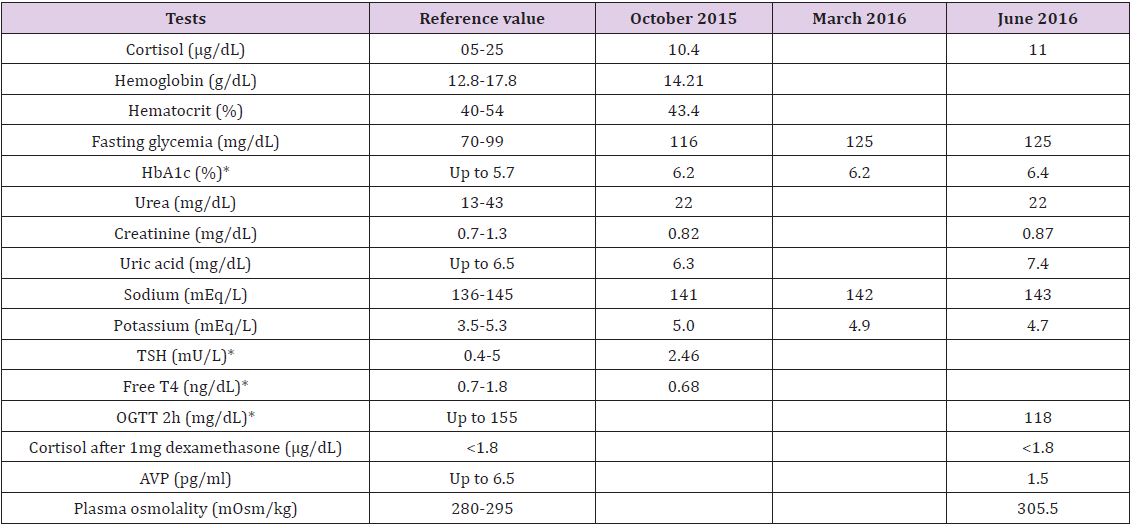
Note: *HbA1c: glycosylated hemoglobin, TSH: thyroid-stimulating hormone, T4: thyroxine, OGTT: oral glucose tolerance test, AVP: arginine vasopressin.
Table 2: Results of Urine Tests.
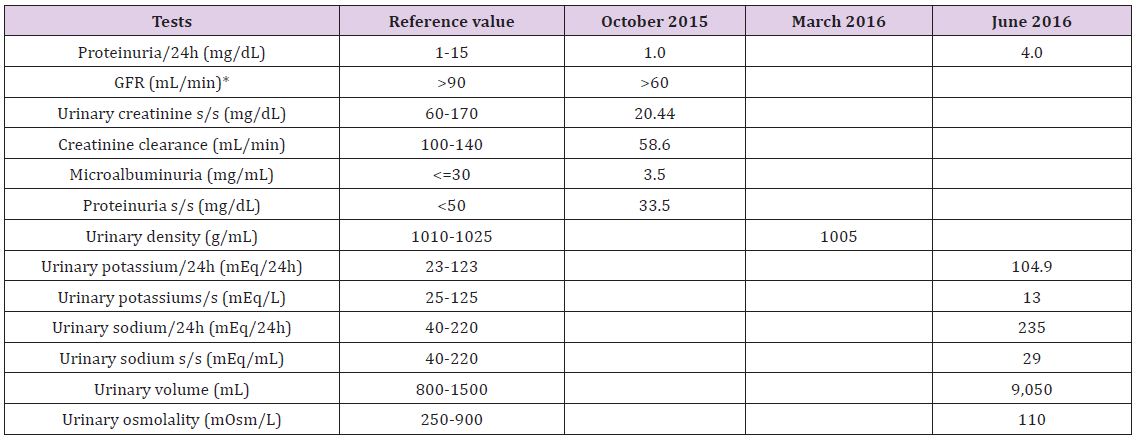
Note: *GFR: glomerular filtration rate,s/s (single sample).
On clinical examination, hepatic steatosis was seen on total abdominal ultrasonography; fundus oculi were normal; arterial pressure was 140/90mmHg; weight was 91kg; abdominal circumference was 111cm; and BMI was 30.7kg/m2. Adrenal insufficiency was discounted. The patient was diagnosed with metabolic syndromeon the basis of systemic arterial hypertension, dyslipidemia, and increase in abdominal circumference. Medications were maintained, and orlistat (120 mg twice a day) was prescribed, in addition to a 1500kcal diet. At the patient’s next visit on November 27th, 2015, 2 months later, his weight loss was 5.5kg, BMI was 29.4kg/ m2, and his abdominal circumference had reduced to 103cm. DI was investigated due to urine volume of 3350mL/day. He had already stopped taking metformin. Orlistat was stopped and further tests were requested. Submitted tests performed on October 08th, 2015.
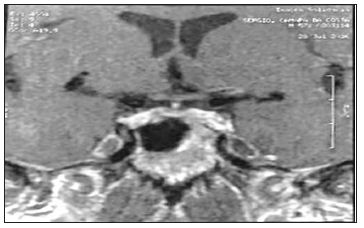
On March 21th, 2016, the patient returned to the clinic complaining of intense thirst and urinating excessively. His arterial blood pressure was 160/110mmHg and his weight 86.9 kg. He no longer reported hypoglycemia. Plasma osmolality, which was calculated using the equation 2×(Na+K)+(glycemia/18), was 300.74mmol/L (reference values 280-285mmol/L). Further test results ashown in (Tables1 & 2). On June 28th, 2016, he attended with the results of tests carried out on June 1, 2016 to confirm the diagnosis is shown in Table 1. The patient ingested 7250mL/dayof fluid and urinated 7100mL/day. We hypothesized that central DI (CDI) was the most probable diagnosis, and conducted further tests onJune 28, 2016 (see (Tables 1 & 2). Magnetic resonance imaging (MRI) of the sella turcica showed a thickened pituitary stalk andconfirmed the diagnosis of CDI (Figures 1 and 2). AVP replacement therapy was prescribed. T1-weighted sagittal MRI showed a small reduction of hyper intense neurohypophysis signal.
Figure 1: Magnetic Resonance Imaging (MRI) of the sella turcica. Note: T1-weighted sagittal MRI showed a small reduction of hyper intense neurohypophysis signal.
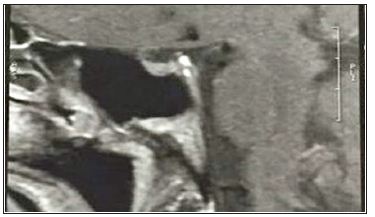
Figure 2: Magnetic Resonance Imaging (MRI) of the sella turcica Note: T1-weighted axial MRI showed a thickened pituitary stalk.
DI is characterized by polyuria, the excessive secretion of diluted urine, in adults of volumes greater than 3L/day. DI may result from disorders related to AVP. Insufficient synthesis of AVP is a sign of CDI, neurogenic diabetes, or vasopressin-responsivediabetes. Further, a decrease in renal sensitivity to AVP is a sign of NDI or non-AVP-responsive DI [5,6]. CDI is a heterogeneous condition characterized by polyuria and polydipsia due to deficiency of AVP. In some patients, it is caused by destruction or degeneration of neurons originating in the supraoptic and paraventricular nuclei of the hypothalamus. Known causes of such lesions include germinoma, craniopharyngioma, Langerhans cell hystiocytosis, inflammatory, autoimmune, or vascular disorders, trauma resulting from surgery or accident and, in rare cases, inherited genetic defects in synthesis of AVP. However, most cases are considered idiopathic [6]. Maintaining the water balance in healthy humans is due to three factors: thirst, AVP, and the kidneys [7]. Plasma osmolality is maintained within a narrow range varying between 285 and 295mOsm/kg, and this depends on the balance between water input (controlled by the physiological sensation of thirst) and renal water secretion (controlled by AVP secretion and action). Physiologically, plasma osmolality is the main thirst regulator.
When an increase in plasma osmolality is detected by osmotically sensitive neurons (osmoreceptors) located in the anterior hypothalamus, AVP is liberated. This increases resorption of water in the renal collecting tubules, stimulating thirst. The increase in ingested water reduces plasma osmolality to levels at which the control of urine production mediated by the increase in AVP maintainsosmolality within its normal range [5,6,8]. CDI may becongenital, and may have a genetic cause. However, it is acquired in 95% of cases [9,10]. Familial CDI is the most common genetic form. It has a similar incidence in both sexes and most frequently presents as a progressive deficiency in the secretion of AVP. Acquired causes include any surgical, traumatic, ischemic, infectious, or infiltrative lesion or tumor in the hypothalamic-hypophyseal region leading to destruction of AVP-producing neurons or impeding transportation of this hormone by the pituitary stalk [5,10,11]. Several tumors of the central nervous system may cause DI, but craniopharyngioma is the main etiology. Other tumors that may cause DI are germinoma, meningioma, glioma, and astrocytoma [5,6,10,12,13]. Cranial trauma resulting in pituitary shaft contusion may be a cause of DI [14]. Transitory DI is the main complication of transsphenoidal surgery [15]. However, craniotomies in which neoplasms are resected often result in permanent DI [5]. The main infectious causes of DI are meningitis, encephalitis, sellar- and super-sellartuberculomas, and infection with cytomegalovirus or neurosyphilis [5,6,10,16,17]. Other causes may be lymphocytic hypophysitis, aneurysms, thromboses, Sheehan syndrome, and multiple sclerosis [5,6,18-20].
The diagnosis of DI is made on the basis of the patient’s clinical history, laboratory tests, and imagingresults [21].The main clinical symptoms of DI are polyuria and polydipsia, which manifest regardless of thetime period.The laboratory findings that indicate DI are persistent hyposthenuria, with specific density below 1010g/mL, and urinary osmolality (UOSM) below 300mOsm/kg. Plasma osmolality (POSM) is normal or slightly high, according to the patient’s thirst and the amount of water ingested. Determining POSM or basal serum sodium is not diagnostically useful because values in patients with CDI, NDI and primary polydipsia (PP) are normal and may overlap. Nonetheless, if these values are clearly above normal levels (POSM>295 mOsm/kg and Na>143mEq/L) in conditions where ingestion of water is unrestricted, a diagnosis of PP is excluded. Consequently, a differential diagnosis must be made between CDI and serious NDI [5,6,10,11]. For a differential diagnosis, DDAVP (10μg subcutaneously) is administered, and after one and two hours assessment, non-concentrated urine indicates NDI, and concentrated urine, CDI [21]. In cases where a cause for polyuria cannot be found, water deprivation is advised [21]. Water deprivation is carried out in two stages [22]: First, during a period of water fasting, the patient’s weight (after hourly urine elimination) and urine osmolality are checked every hour, and plasma osmolality and serum sodium every two hours [22].
Figure 3: Water Deprivation Test. Note: a. DDAVP:desmopressin acetate b. OsmU: Urinary osmolality c. CDI: central diabetes insipidus d. NDI: nephrogenic diabetes insipidus
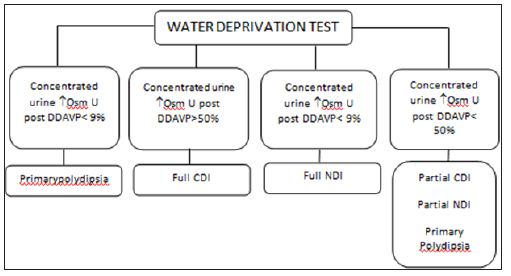
Second, 40 μg of intranasal desmopressin is administered, free fluids given orally, and plasma osmolality and urinary osmolality checked one and two hours, respectively, after administration of desmopressin [21]. This is illustrated in (Figure 3). In CDI cases, the gold standard imaging techniqueto search for tumors or other pathologies in the hypothalamic-hypophyseal region is MRI. MRI may also reveal expanding lesions in the sella turcica region (e.g., tumors, abscesses, infiltrative diseases, hypophysitis, etc.) and thickening of the pituitary stalk [23]. In up to 80% of CDI cases, the posterior pituitary bright spot is visualized on MRI. However, this is also observed in up to 20% of normal people or people who have NDI, which may be secondary to depletion of AVP caused by chronic, excessive secretion. Patients with a diagnosis of idiopathic CDI with no evidence of tumors, infiltrative diseases, infectious diseases or metastatic diseases on initial MRI images should have subsequent MRIs because, in some cases, tumors may appear a few years after diagnosis [24].
Differential diagnoses could be PP, CDI or NDI [21]. PP includes psychiatric diseases, absence of nocturia, or episodic polyuria. A urinary volume greater than 18L is highly suggestive of PP. However, most CDI patients have moderate dehydration, a decrease in urine volume (6 to 12L), and a decrease in the glomerular filtration rate [21]. In CDI, patients develop polyuria after cranial trauma or neurosurgery; it is also found in patients with clinical evidence of sella turcica or metastatic tumors (lung and breast), granulomatous diseases (Langerhans cell histiocytosis, sarcoidosis, and tuberculosis), and infectious diseases (pituitary abscesses, meningitis, encephalitis, and neurosyphilis) [25]. NDI is characterized by the kidney’s inabilityto concentrate urine despite normal concentrations of AVP. It may be caused by hydroelectrolytic disorders (hypopotassemia, hypercalcemia), chronic renal disease or ingestion of DI-inducing substances (lithium, amphotericin B, methotrexate, etc.). A rare form is familial CDI, caused by mutation of the receptor gene V2 [5,6,10,11].
Specific treatment for CDI is AVP replacement. The medicine of choice is Desmopressin, an AVP-analog with minimal pressor effect, greater diuretic activity and extended half-life, with a 6 to 24 h duration of action [26].
Medicationformats aretablets (0.1 and 0.2mg), nasal spray (0.1 mg/mL), nasal solution (0.1 mg/mL), and injectable solution (4 μg/ mL). The best therapeutic form will depend on the physician and the particular characteristics of the patient. The number of doses for optimal control of the disease will also depend on the patient’s clinical characteristics [26-28].
In cases of CDI with mild or moderate polyuria, chlorpropamide (CLORP) (125 to 500mg/day) may be trialed. Doses greater than 500mg/day do not produce any greater therapeutic effect and may potentiate the action of AVP. CLORP is contraindicated in pregnant women and children due to risk of hypoglycemia [5,10,24,29]. Thiazide diuretics such as hydrochlorothiazide (50 to 100mg/day) may alternatively be trialed if CLORP produces anun satisfactory response. Indapamide has a similar effect [5,6,10,11]. Carbamazepine (200 to 400 mg/day) is indicated in patients unresponsive to the previous options. It stimulates the secretion of AVP by hypothalamic neurons and, in some cases, increases renal sensitivity to the hormone. The use of carbamazepine is limited by side effects including skin rashes, diplopia, blurred vision, drowsiness, nausea, vomiting, and ataxia [6,10-12].
Diabetes Insipidusrepresentsa small part of diabetes cases in our population. DM2 account for the great majority of cases of Diabetes [1]. Complaints of polydipsia and polyuria are common in patients with uncontrolled DM2. In our patient’s case, his complaints at the initial consultations suggested likely uncontrolled DM2. However, continuous monitoring of capillary glycemia and glycosylated hemoglobin showed that the patient did not have significant enough discomfort to reportfurther symptoms. Other important causes of polyuria and polydipsia are DI and PP. Measuring urine volume for 24 h and urine density via a simple urine test were key to diagnosing the type of diabetes. The patient spent two years receiving treatment for DM2, and experienced frequent episodes of hypoglycemia. In such cases, it is also important to exclude adrenal insufficiency; the basal cortisol level was greater than 10μg/L, which helped exclude adrenal insufficiency. We did not investigate possible insulinoma because there was no classic Whipple’s triad of hypoglycemic symptoms and no significant weight gain, both of which are typical of insulinomas. Differential analysis depends on exclusion criteria that compare symptoms and laboratory anomalies to diseases that are similar to NDI.
Urine density analysis is a differential criterion. In DM2, this is high, above 1020 mmol/L, whereas in neurogenic DI urine is hypotonic, i.e., with a density less than 1010mmol/L [30]. In our patient’s case, the symptoms of polyuria and polydipsia, alteration in arterial pressure, being overweight and slight hyperglycemia aroused suspicion of DM2, so treatment was initiated. The patient experienced side effects due to the use of oral hyperglycemic agents and rigorous diet. When he started treatment at our medical clinic, his urinary density of 1005 g/mLindicated DI. The plasma sodium level of 143mEq/L, characteristic of hypernatremia, wasan important differential indicator ofCDI; in hypernatremia, plasma sodium levels are greater than 135 mEq/L, whereas in NDI, hyponatremia is observed [31]. Another differential parameter was plasma osmolality, calculated by the formula POSM=2×(Na+K)+(glycemia/18), which, in our patient’s case was 300.74mOsm/kg. When osmolality is greater than 282mOsm/kg, the thirst mechanism is activated, and the POSMreference value varies between 280 and 295 mOsm/kg. POSM is a measurement that indirectly assesses a patient’s degree of hydration [19]. The patient’s urinary osmolality was 110mOsm/kg, and, in central DI, the reference value is below 300mOsm/kg. PP is another possible differential diagnosis. Nonetheless, an important aspect encountered by the patient was intense nocturia, which harmed his sleep quality, a complaint that is infrequent in PP patients.
Nocturia is the first symptom to manifest in DI. However, most patients do not complain about it until diuresis exceeds 4000mL/ day [20]. CDI is characterized by polyuria and polydipsia, which exacerbates excessive nocturia, leading to alteration of sleep patterns, fatigue and irritability [21]. The patient ingested 7250mL of fluid and had diuresis of 7500mL. Therefore, diuresis was 87.2mL/kg, which falls within the definition of polyuria, a volume of urine greater than 45-50mL/kg in 24h. Total AVP deficiency would lead to diuresis greater than 18,000mL/24h [22]. Slightly elevated diuresis suggests partial AVP deficiency, which may explain why the AVP value in this case was 1.5pg/mL. The reference value for CDI is below 1.0pg/dL [6,11]. The next step was cranial MRIto confirm disease etiology. The most common hypophyseal alterations in DI are thickening of the pituitary stalk and fading of the posterior pituitary bright spot visualized in a T1 weighted image on MRI. The thickening of the pituitary stalk confirmed the diagnosis, and AVP replacement treatment was initiated [6, 11] (Figures 1 and 2).
Today, most of CDI previously classified as idiopathic have been now verified as an autoimmune etiology. Some features of the present case suggest an autoimmune pathogenesis. The imaging findings on MRI, showing a thickened pituitary stalk and a reduction of hyperintense neurohypophysis signal, favor an autoimmune CDI caused by a lymphocytic infundibulo-neurohypohysitis. To search for the presence of antihypothalamus and antipituitary antibodies in patient’s serum could have contributed to clarify these aspects, unfortunately the patient did not perform these exams.
This case report highlights the differential diagnoses and possible causes of DI. In the case, we report small abnormalities in laboratory tests, which initially were not noticed by nonspecialists,making it difficult to reach anearly diagnosis. Moreover, the frequency of radiological abnormalities in patients with CDI is ill defined. After clinical investigation, CDI, also known as neurogenic DI, was diagnosed based on abnormalitiesin laboratory tests and morphologic characteristics of the neurohypophysis on MRI. Today, CDI cases previously classified as idiopathic have been now verified to have an autoimmune pathogenesis. CDI is a disease that causes many problems to those who suffer from it, including a reduction in their quality of life. For this reason, early diagnosis based on differentiation fromPP, careful note of small abnormalities in laboratory tests, and particularly careful patient historyare key points to commencement of early treatment in order torestore these patients’ well-being and quality of life.

























