info@biomedres.us
+1 (502) 904-2126
One Westbrook Corporate Center, Suite 300, Westchester, IL 60154, USA
Site Map
Received: November 03, 2017; Published: November 15, 2017
Corresponding author: Sherin DR, Post Doctoral Fellow, Centre for Computational Modeling and Data Engineering, Indian Institute of Information Technology and Management-Kerala, Thiruvananthapuram, Kerala, india-695581, Trivandrum, Kerala-695581, India
DOI: 10.26717/BJSTR.2017.01.000533
Breast cancer is one of the major causes of mortality in women. The estrogen receptor becomes positive in such patients. Many of the currently available anti breast cancer drugs produces severe side effects. It demands natural products with low toxicity to fight against estrogen receptor. In this point, we tried to establish antiestrogen potential of some natural flavonoids from the bark of Saraca asoca. The vitality of these drugs is well described in terms of pharmacokinetic parameters and molecular orbital analysis. In the light of molecular simulation studies we reports a set of lead compounds having natural origin in the treatment of breast cancer.
Keywords: Breast cancer; Estrogen receptor; Flavanoids; Catechins; Saraca asoca
Breast cancer is one of the most widely affected malignancies in women, with 2,52,710 new case reports and death of 40,610 in 2016 [1]. It is well known that breast cancer is associated with steroid hormone estrogen receptor (ER) and it becomes the best target for the development of therapeutics [2]. Tamoxifen is one of the commonly used antiestrogens in the treatment of breast cancer, which act as a selective estrogen receptor modulator (SERM) [3]. But it harmfully affects oncologic, musculoskeletal, metabolic, hepatic, cardiovascular and even nervous systems [4- 9]. Hence it is necessary to suggest natural medications which are of low toxicity than tamoxifen for the metastasis in breast cancer. Saraca asoca possess various medicinal values, its stem bark is the principal constituent of major ayurvedic preparations for leucorrhoea, haematuria, menorrhagia and other diseases of the female genitourinary system [10]. Saraca asoca was extensively interpreted for its anti platelet aggregation, cytotoxic, anti inflammatory anti acne and anti oxidant activities [11-15]. The dried bark extract of Ashoka mainly contains lignan glycosides and flavonoids [16]. The flavanoids includes catechin monomers such as catechin, epicatechin, epigallocatechin, leococyanidin, gallocatechin and leucopelargonidin [17]. The importance of three dominant polyphenolic catechins-epigallocatechin, epigallocatechin gallate and epicatechin gallate in breast cancer cell proliferation was reported in 2002 [18]. Ryoko et al. studied the simultaneous effect of tea catechins in anti estrogenic activity [19]. As part of our interst in the development natural anticancer therapeutics, we are concentrating to elaborate the ER inhibitory activities of monomeric catechins from Saraca asoca and defined it as an ideal medication for breast cancer based molecular simulation studies.
The crystal structure of ER (PDB ID: 3OS8) was retrieved from RCSB Protein Data Bank [20]. The X-ray crystal structure has a resolution of 2.03Å and it contains 258 amino acids. It was cleaned and prepared for docking studies using Protein preparation wizard of Schrodinger suite 2017-2 [21]. The different conformers of the ligands-catechin(+) (1), catechin(-) (2), epicatechin(+) (3), epicatechin(-) (4), epigallocatechin(+) (5), epigallocatechin(-) (6), leucocyanidin(+) (7), gallocatechin(+) (8), gallocatechin(-) (9) and leucopelargonidin(+) (10) were prepared by ligprep and their absorption, distribution, metabolism, excretion and toxicity(ADME/T) were described by QikProp analysis. The prepared ligands were docked against the 3OS8 using Glide programme of Schrodinger suite and the compounds were screened based on D-score and G-score values.
The ten monomeric flavanoids selected were optimized at B3LYP/6-311++G**, the prepared ligands were docked against the grid generated around the highest scored site of 3OS8. From the binding scores, all of the ligands show excellent D-Score/G-score of less than -10.8kcal/mol, confirms the strong binding capacityof the ligands inside the active site of 3OS8 (Table 1). The scores are better than the values reported for flavanoids by Suganya [22]. The drug ability parameters such as #stars, CNS, FISA, HBA, HBD, QPlogPo/w, QPlogS and QPlogKhsa are well within the acceptable range with minimum deviation from Lipinski rule of five (Ro5). The low molecular weight of 290-306 range increases the cell permeability of the ligands with blood brain barrier permeability of -2. The predicted skin permeability and oral absorptions are also in the recommended range. Most of the clinically successful drugs in the market have had the tendency to inhibit hERG with QPlogHERG value less than -5, but our ligands are exception to this threat. The hydrophobic and π-components of solvent accessible surface areas are matched well with the site volume of 204.77Å3 of 3OS8. The globularity descriptor value of 0.86 shows the spherical character of the ligands which may facilitate the binding of them in the binding pocket (Table 1).
Table 1: D-score/G-score and pharmacokinetic parameters for 1-10.
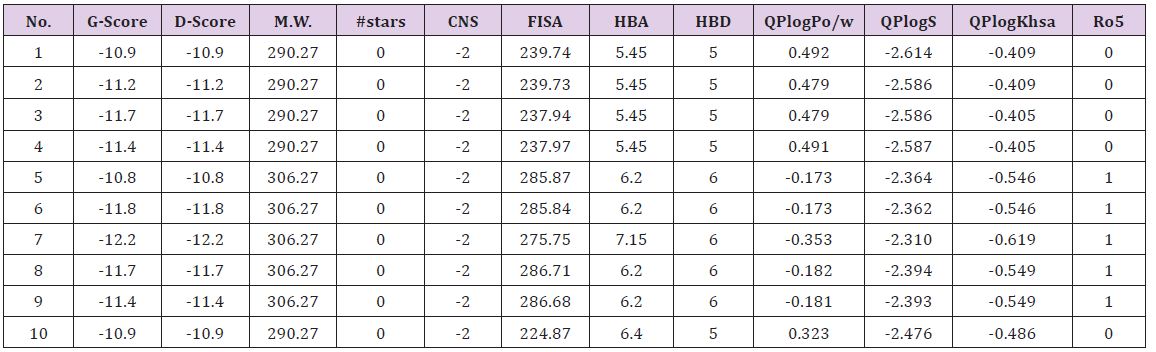
M.W.(Molecular Weight):130.0-725.0; #stars (few stars-more drug-like): 0-5; CNS (Central Nervous System activity): -2 to +2; FISA (Hydrophilic component of total solvent accessable area): 7.0-333.0; HBA(Hydrogen bond acceptor): 2.0-20.0; HBD(hydrogen bond donor): 0.0-6.0; QPlogPo/w (octanol/water partition coefficient): -2.0-6.5; QPlogS (Aqueous solubility): -6.5-0.5; QPlogKhsa (binding to human serum albumin): -1.5-1.5; Ro5 (Number of violations of Lipinski’s rule of five): maximum is 4.
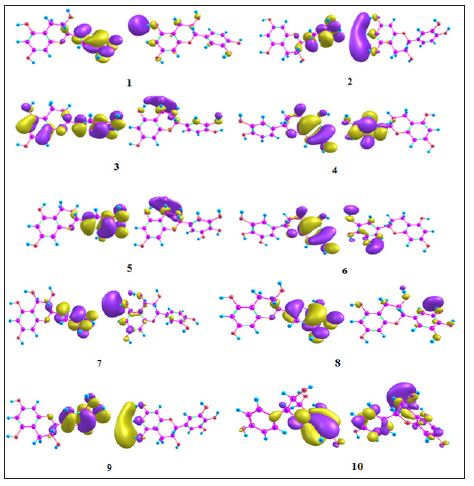
The close observation of interaction diagram Figure 1 shows that all of the ligands almost closely packed in the binding site which gives the appreciable values. All the ligands except 5 forms H-bond with negatively charged Glu353 while the eight ligands except 5 and 9 have H-bond with positively charged Arg394 and 5 forms only one bond with His524. The HOMO-LUMO diagrams Figure 2 depict the electronic transition from HOMO to LUMO by a distance of 5eV, which is favorable for interaction of terminal hydroxyl groups with the aminoacid residues. The electron density in the HOMOs are concentrated on the benzene ring on 1, 2, 5, 6, 7, 8 and 9 while it is on chromenyl moiety in the case of 4 and 10. The hydrogen of OH from the corresponding rings is in strong interaction with the negatively charged glutamic acid. At the same time the oxygen’s of OH have the tendency to interact with positively charged arginine. These interactions from hydroxyl groups cause the strong binding of the ligands inside the binding pocket (Figures 1 & 2).
Figure 1: Interaction diagrams of 7, 6 and 3.
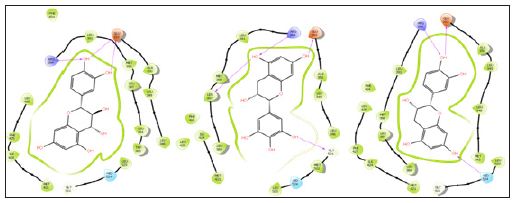
Figure 2: HOMO-LUMO diagrams of 1-10.
We are able to suggest a set of lead molecules from the natural source, bark of Saraca asoca. The interaction diagram shows the strong interactions of hydroxyl groups with negatively charged glutamic acid and positively charged arginine, which favors the strong binding. The molecular orbital analysis clearly tells the delocalization of electron density in the benzenyl and chromenyl hydroxyl groups. The pharmacokinetic parameters strongly support the druggability of these ligands. This study is expected to pave the way of generating new chemotherapeutics in the treatment of metastasis in breast cancer with low toxic effects.
The authors thank IIITMK, Trivandrum for providing research facilities. SDR thanks KSCSTE for Post Doctoral Fellowship.

























