info@biomedres.us
+1 (502) 904-2126
One Westbrook Corporate Center, Suite 300, Westchester, IL 60154, USA
Site Map
Received: October 14, 2017; Published: October 25, 2017
Corresponding author: Priyambada Pandey, BN Institute of Pharmaceutical Sciences, Sewashram Choraya, Udaipur, India
DOI: 10.26717/BJSTR.2017.01.000463
Quality is always an imperative prerequisite when consider any product. Therefore, drugs must be manufactured to the highest quality levels. Validation is a part of the quality assurance program and is fundamental to an efficient production operation for building quality into the products. Validation of the individual steps of the processes is called the process validation. Process validation involves the collection and evaluation of data, from the process design stage throughout production, that establish scientific evidence that a process is capable of consistently delivering a quality drug substance. The goal of the validation is to ensure that quality is built into the system at every step, and not just tested for at the end. It involves the collection and evaluation of data, from the process design stage throughout production, that establish scientific evidence that a process is capable of consistently delivering a quality drug substance. Process validation is an integral part of Quality Assurance as per cGMP. Validation and quality assurance will go hand in hand, ensuring the thorough quality for the products. Hence, an emphasis made on to review that gives a detailed, overview of validation. According to GMP, validation studies are required to be done as per predefined protocols. Different dosage forms have different validation protocols. The purpose of this work is to present an introduction and general overview on process validation of pharmaceutical manufacturing process with special reference to the requirements stipulated by the US Food and Drug Administration (FDA) of Solids (tablets and capsules), liquids and semisolids.
Keywords: Quality; Validation; Dosages form; CGMP
The main goal of any pharmaceutical plant is to produce products of required quality consistently, at the lowest cost possible. Validation has been an important process in pharmaceutical industries for a long time but it has gained greater emphasis in recent years due to industry’s greater interest on assurance of quality and productivity improvement. For an efficient production operation, there is a greater need of an efficient validation team [1]. The concept of validation was first proposed by two Food and Drug Administration (FDA) officials, Ted Byers and Bud Loftus, in the mid 1970’s in order to improve the quality of pharmaceuticals. The prime focus of validation is on ensuring if the quality is built into the system at every step, and not just tested for at the end [2]. Validation is documented act of proving that any procedure, process, equipment, material, activity or system actually leads to the expected results [3].
Process validation provides the flexibility and constraints in the production process controls in the achievement of desirable qualities in the drug product while preventing undesirable attributes [4]. USFDA defined process validation as “establishing documented evidence which provides high degree of assurance that a specific process will consistently produce a product meeting its pre determined specifications and quality characteristics.” [5,6] (Figure 1).
Figure 1: Flow chart describing validation process.
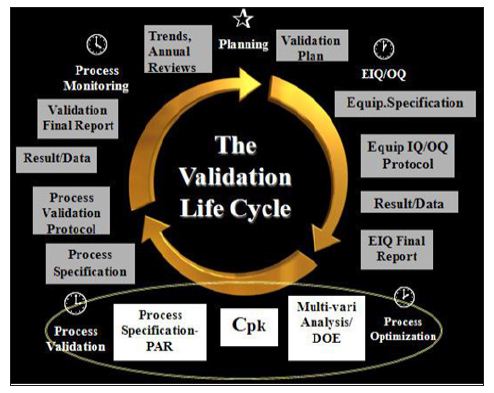
a) Prospective Process Validation: In prospective process validation, the experimental plan known as validation protocol (following completion of the qualification trials) is prepared before the process is used for commercial use. In order to produce support data for validation there is requirement of some degree of prospective experimentation.
b) Concurrent Process Validation: The concurrent process validation establishes documented evidence that the process is in a state of control during the actual execution of the process. The in-process testing and/or monitoring of critical operations during the manufacture of each production batch is done for concurrent process validation.
c) Retrospective Process Validation: When validation is based on the historic data taken from the records of the completed production batches and used as a documented evidence for stating that the process has been in a state of control comes under retrospective process validation.
d) Revalidation: Revalidation ensures that changes in the process and/or in the processing environment, whether intentional or unintentional, do not negatively affect process characteristics and product quality attributes.
Revalidation can be sub-divided into two categories:
i. Revalidation after any change having a bearing on product quality.
ii. Periodic revalidation carried out at scheduled intervals (Figure 2).
Figure 2: Stages of Process Validation.
a) Quality of product is assured
b) Optimization of the process
c) The cost of Quality of products is reduced
d) The market recalls of products is minimized
e) The process is under control and detailed study is possible
i. Validation master plan
ii. Validation protocol
iii. Validation report
iv. SOPs
i. Validation Master Plan [9]: A validation master plan is a summary of entire philosophy, intentions and approaches to be used for establishing performance adequacy of the company. The management must agree upon the validation master plan. The validation master plan provides an outlook of the overall validation operation, its organizational structure, its content and planning.
ii. Validation Protocol [10]: Validation protocol is a plan of actions stating how process validation will be done, it specifies who will conduct the various tasks and defines the testing parameters, sampling plans, testing methods and specifications. It also specifies product characteristics and equipments to be used. It must state the minimum number of batches that can be used for validation studies, it must specify the acceptance criteria and who will sign \ approve \ disapprove the conclusions obtained from such a study.
The validation protocol should be numbered, signed and dated, and should contain as a minimum the following information:
a. Title
b. Objective & Scope
c. Responsibility
d. Protocol Approval
e. Validation Team
f. Product Composition
g. Process Flow Chart
h. Manufacturing Process
i. Review of Equipments / Utilities
j. Review of Raw Materials and Packing Materials Review of Analytical and Batch Manufacturing Records
k. Review of Batch Quantities for Validation (Raw Materials)
l. Review of Batch Quantities for Validation (Packing Materials)
m. HSE Requirements
n. Review of Process Parameters Validation Procedure
o. Sampling Location
p. Documentation
q. Acceptance Criteria
r. Summary
s. Conclusion
iii. Validation Report [11]
Standard format of validation report
A. Executive summary
B. Discussion
C. Conclusions & recommendation
D. List of attachment
The above topics should be represented in the series in which they appear in the protocol and if the protocol is deviated it should be explained with justification. The report must be signed & dated by designated representatives of each unit.
iv. SOP (Standard Operating Procedure) [12]
The general format of the SOPs involves:
A. Title
B. Code
C. Objective
D. Scope
E. Definitions
F. Description
G. Safety
H. Documentation
I. Effective date, review date, version number.
J. Footer: Prepared By, Reviewed By, Approved By, Authorized By.
K. References
A tablet is a most known solid pharmaceutical dosages form and comprises of a mixture of active substances and suitable excipients. Binders, glidants, lubricants etc are some the popularly used excipients in the tablets. The excipients are used for different purposes in the tabletting; like disintegrants used to enhance the breakdown, glidants used to increase the flow of the powder, flavouring agents to impart different flavours in the tablets. The knowledge of stepwise manufacturing process of any dosages form is a must for validating any process. It helps in determining the critical areas which need special consideration in terms of causing problems during the process.
Mixing is one of the most critical step and used at various stages during manufacturing of tablets. Materials with like physical properties can easily form a uniform mix or blend and not segregate as soon as materials with large differences.
Parameters to consider:
a) Mixing Or Blending Technique: The techniques like Diffusion (tumble), convection (planetary or high intensity), or pneumatic (fluid bed) are used to mix or blend materials. The choice of technique depends on whether the drug and excipients are mixed for a direct compression formulation or for adding the lubricant (e.g., magnesium stearate) to the granulation.
b) Mixing or Blending Speed: Mixing the drug and excipient requires more intense mixing than adding the lubricant to the final blend.
c) Mixing or blending time: The mixing or blending time of the product will be dependent on the mixing or blending technique and speed.
d) Drug uniformity: The test for content uniformity is usually performed to estimate the uniformity of drug throughout the mix or blend.
e) Excipient uniformity: Besides drug uniformity, excipients uniformity is also necessary in the granulation or blend. Two key excipients are:
f) Lubricant: Uneven distribution of the lubricant can result in picking and sticky problems during compression. It can also lead to tablet performance problems (low dissolution due to excessive lubricant in some tablets).
g) Color: The colorant(s) need(s) to be evenly distributed in the mixture so that the tablets have a uniform appearance (e.g., color, hue, and intensity).
h) Equipment capacity/load: The bulk density of materials or granules will affect the capacity of the equipment. Undercharging or overcharging a blender can result in poor drug or tablet lubricant distribution.
Wet granulation parameters to be considered during development and validation are:
a) Binder Addition: Adding the binder dry avoids the need to determine the optimal binder concentration and a separate manufacture for the binder solution.
b) Binder Concentration: The optimal binder concentration will need to be determined for the formulation. If the binder is to be sprayed, the binder solution needs to be dilute enough so that it can be pumped through the spray nozzle. It should also be sufficiently concentrated to form granules without over wetting the materials.
c) Amount of Binder Solution/Granulating Solvent: Too much binder or solvent solution will over wet the materials and prolong the drying time. The amount of binder solution is related to the binder concentration.
d) Binder Solution/Granulating Solvent Addition Rate: The rate or rate range at which the binder solution or granulating solvent can be added to the materials should be defined properly.
e) Mixing Time: Granulations that are not mixed long enough can form incomplete or weak granules. These granules may have poor flow and compression properties. On the other hand, over mixing the granulation can lead to harder granules and a lower dissolution rate.
Sometimes wet milling of granules is needed before subjecting it for drying to efficiently dry them.
Factors to consider are:
a) Equipment Size And Capacity: The mill should be large enough to de lump the entire batch within a reasonable time period to minimize manufacturing time and prevent the material from drying during this operation.
b) Screen Size: The screen needs to be small enough to de lump the material, but not too small to cause excessive heating of the mill, resulting in drying of the granulation.
c) Mill Speed: The speed should be sufficient to efficiently de lump the material without straining the equipment.
d) Feed Rate: The feed rate of the wet granulation is interrelated to screen size and mill size and speed.
The type of drying technique (e.g., tray, fluid bed, and microwave) required for the formulation needs to be determined and justified. The type of technique may be dependent on such factors as drug or formulation properties and equipment availability. Changing dryer techniques could affect such tablet properties as hardness, disintegration, dissolution, and stability.
The optimal moisture content of the dried granulation needs to be determined.
i. High moisture content can result in
a) Tablet picking or sticking to tablet punch surfaces and
b) Poor chemical stability as a result of hydrolysis.
ii. An over dried granulation could result in poor hardness and friability.
Parameters to consider are:
A. Inlet/Outlet Temperature: The inlet temperature is the temperature of the incoming air to the dryer, while the outlet temperature is the temperature leaving the unit. The inlet temperature is critical to the drying efficiency of the granulation and should be set high enough to maximize drying without affecting the chemical/physical stability of the granulation. The outlet temperature is an indicator of the granulation temperature and will increase toward the inlet temperature as the moisture content of the granulation decreases (evaporisation rate).
B. Airflow: There should be sufficient airflow to ensure removal of moisture laden air from the wet granulation. Insufficient air flow could prolong drying and affect the chemical stability of the drug.
C. Moisture Uniformity: The moisture content could vary within the granulation
D. Equipment Capability/Capacity: The load that can be efficiently dried within the unit needs to be known.
The milling operation will reduce the particle size of the dried granulation. The resultant particle size distribution will affect such material properties as flow, compressibility, disintegration, and dissolution. An optimal particle size/size distribution for the formulation will need to be determined. Factors to consider in dry milling are same as that of wet milling.
Lubricants are added in order to remove the problem of sticking and picking in the tablets.
a) Selection of Lubricant: Grade of the lubricant used and compatibility with other ingredients should be studied thoroughly and then the appropriate one must be chosen.
b) Amount of Lubricant Added: How much lubricant is required? Too much lubricant will form hydrophobic layer on the tablet resulting in dissolution problems.
c) Mixing Time: The optimum mixing time must be decided on proper trial of batches because if not mixed long enough form problems like chipping, capping, etc.
Compression is a critical step in the production of a tablet dosage form. As for the compressibility properties of the formulation, it should be examined on an instrumented tablet press. Factors to consider during compression are as follows:
A. Tooling: The shape, size, and concavity of the tooling should be examined based on the formulation properties and commercial specifications.
B. Compression speed: The formulation should be compressed at a wide range of compression speeds to determine the operating range of the compressor.
C. Compression/ejection force: The compression profile for the tablet formulation will need to be determined to establish the optimal compression force to obtain the desired tablet hardness.
The following in-process tests should be examined during the compression stage:
I. Appearance
II. Hardness
III. Tablet weight
IV. Friability
V. Disintegration
VI. Weight uniformity
VII. Tablet Coating
Film coating has been the most common technique over recent years and will be the focus of this section.
Key areas to consider for tablet coating include the following:
a) Tablet Properties: Tablet properties such as hardness, shape, and intagliation (if required) are important to obtain a good film-coated tablet. The tablet needs to be hard enough to withstand the coating process
b) Equipment Type: The type of coater will need to be selected. Conventional or perforated pan and fluid bed coaters are potential options.
c) Coater Load:Having too large a pan load could cause attrition of the tablets because of the overall tablet weight in the coater. In the case of a fluid bed coater, there may not be sufficient airflow to fluidize the tablets.
d) Pan Speed: This will be interrelated to other coating parameters, such as inlet temperature, spray rate, and flow rate.
ve) Spray Guns: The number and types of guns should be determined in order to efficiently coat the tablets.
f) Application/Spray Rate: The optimal application/spray rate should be determined. Spraying too fast will cause the tablets to become over wet, resulting in clumping of tablets and possible dissolution of the tablet surface. Spraying too slowly will cause the coating materials to dry prior to adhesion to the tablets. This will result in a rough tablet surface and poor coating efficiency.
vg) Tablet Flow: The flow or movement of the tablets in the coater should be examined to ensure proper flow. The addition of baffles may be required to provide adequate movement of tablets for tablet coating.
h) Inlet/Outlet Temperature and Airflow: These parameters are interrelated and should be set to ensure that the atomized coating solution reaches the tablet surface and then is quickly dried.
i) Coating Solution: The concentration and viscosity of the coating solution will need to be determined. The solution will need to be sufficiently diluted in order to spray the material on the tablets.
j) Coating Weight: A minimum and maximum coating weight should be established for the tablet.
k) Residual Solvent Level: If solvents are used for tablet coating, the residual solvent level will need to be determined (Table 1).
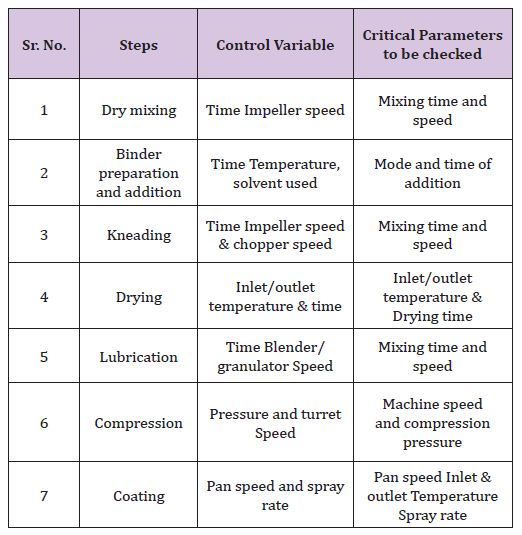
Table 1: Summary table including steps, control variable and critical parameters to be checked in manufacturing of tablets.
Capsules are the solid dosage form in which the drug or the mixture of drug are enclosed in Hard Gelatine Capsule Shells, in soft , soluble shells of gelatine, or in hard or soft shells of any other suitable material, of various shape and capacities. They usually contain a single dose of active ingredients and are intended for oral administration.
They are basically of two types:
a) Hard Gelatin Capsules: It is a solid dosage form in which medications are encapsulated in a two part empty hard gelatin capsule shell. The upper and small part is called ‘CAP’ and the remaining large part is called ‘BODY’. There are 8 different sizes of capsule shell(000,00,0,1,2,3,4,5) with different fill volume. Normally 0 and 2 sized shells are widely used. The shell of hard gelatin capsules basically consists of gelatin, plasticizers and water. Modern day shells may, in addition, consist of preservatives, colours, pacifying agents, flavours, sugars, acids, enteric materials etc.
b) Soft Gelatin Capsules: A soft gel (or a soft gelatin capsule) is a solid capsule (outer shell) surrounding a liquid or semi-solid centre (inner fill). An active ingredient can be incorporated into the outer shell, the inner fill, or both. They process of manufacturing of hard gelatine capsules is same as that of tablets, the only difference is that instead of compressing the granules they are filled in the capsule shell. So the validation process is also the same.
In encapsulation process following additional parameters need to be validated:
A. Capsule Shell Contents
a)Establish the compatibility of the capsule shell and the capsule contents.
b) Determine the hygroscopic nature of the capsule formulation
For example: A hygroscopic formulation (API /excipients) can pull water from the capsule shell, which could affect the API stability.
B. Encapsulation Speed: The formulation should be encapsulated at a wide range of speeds to determine the operating range of the encapsulation.
C. Encapsulation: Encapsulation is a critical step in the production of capsules, similar to the compression for tablet dosage forms, The materials to be encapsulated will need to have good flow properties and a consistent density.
They are liquid preparation in which the drugs are dissolved, suspended or disperse in a suitable vehicle and generally several doses are contained in the bottle.
a) Syrups
b) Solutions
c) Suspension
d) Eye drops
e) Nasal drops etc
a) Particle size and size distribution
b) Particle shape or morphology
c) Microbial count
d) Rheology of solvent or vehicle
e) PH of the solvent or vehicle
a) Appearance
b) pH
c) Viscosity
d) Specific gravity
e) Microbial count
f) Content uniformity
g) Dissolution testing
A. Appearance of the final product indicates the signs of instability and degradation. For e.g. settling of solid particles in case of suspension and turbidity in case of emulsion.
B. Time for mixing or agitation and temperature of process can affect the appearance greatly.
C. PH of aqueous oral formulations should be taken at a given temperature and only after equilibrium has been reached in order to minimize the PH drift.
D. Viscosity affects the settling rate of suspended particles in suspension and coalescence of globules of internal phase in emulsions and also in case of oral solutions it affects the overall appearance of the final product so it must be measured and validated properly.
E. Specific gravity: A decrease in specific gravity of the product like suspensions indicates the presence of air within the structure of the formulation.
F. Microbial count for the final product is essential to validate because by performing microbial count we can select the preservative for the final product storage. There are specifications for each liquid oral product for the bio burden content.
G. Content uniformity affects the dose uniformity in case of multi dose formulations and also affects the homogeneity of the drug within solvent system (Table 2)
Table 2: summary table for validation of liquid dosages form
They are mainly meant for external application e.g. cream, jelly, pastes etc. The consistency of semisolids lies between the solid and liquid and thus the preparation is a challenge for manufacturers.
Process Temperature: It is critical to process at right temperature for successful manufacturing. Too much heating during processing can result in chemical degradation and insufficient heat can lead to batch failures, and excess cooling can result in the precipitation of solubilized ingredients.
Heating and Cooling Rates: The successful consistency of ointments, for example, depends on proper rates of heating and cooling.
a) Heating too slowly can result in poor yields from evaporative loss.
b) Heating too rapidly may burn areas of the batch in contact with the heating surface, which raises the potential for burnt material in the batch.
c) Rapid cooling can result in precipitation/crystallization or increased viscosity.
It is essential to determine the required amount of shear and the optimal mixing methods and speeds. Emulsification typically requires high shear or homogenization to obtain the optimal droplet size and dispersion, while the mixing of a gel may require low shear in order to preserve certain physical characteristics, such as viscosity. Proper mixing speeds must be obtained for each phase at every batch scale. Optimal hydration depends on the amount of shear imparted to initially disperse the polymer into the medium. If the process involves only very low shear mixing, a polymer may never be completely dispersed and hydrated, which may result in an out-of specification viscosity. Equipment, such as a recirculation loop, may also be used to correct uniformity without changing mixing speed or time.
Optimizing mixing time requires identifying the minimum time required for ingredients to dissolve and the maximum mixing time before product failure (e.g., when viscosity begins to drop). For polymeric gels, particularly acrylic acid-based types, over-mixing, especially high shear, can break down the polymer’s structure. In an emulsion, over-mixing may cause the product to separate prematurely, resulting in a drastic decline in viscosity.
Optimizing flow rate involves determining the amount of shear or throughput needed. For example, a water-in oil emulsion may require a slower addition speed than a traditional, oil-in-water emulsion, and the flow rate must be modified appropriately. Care must be taken for any product using a pump. Overhearing can occur if the formulation is pumped too quickly. If pumping is too slow, the formulation will experience extra time in an in-line homogenizer, thus also exposing the formulation to additional shear.
Addition of polymers (Carbomers) and gums (Xanthan) must be performed in a very controlled manner if adding directly to batch. Likewise there are other alternate methods of incorporation are : Eductors such as Tri – Blenders and Quadro Ytron dispersers and preparation of slurry of polymers or gum in a medium of low or no solubility.
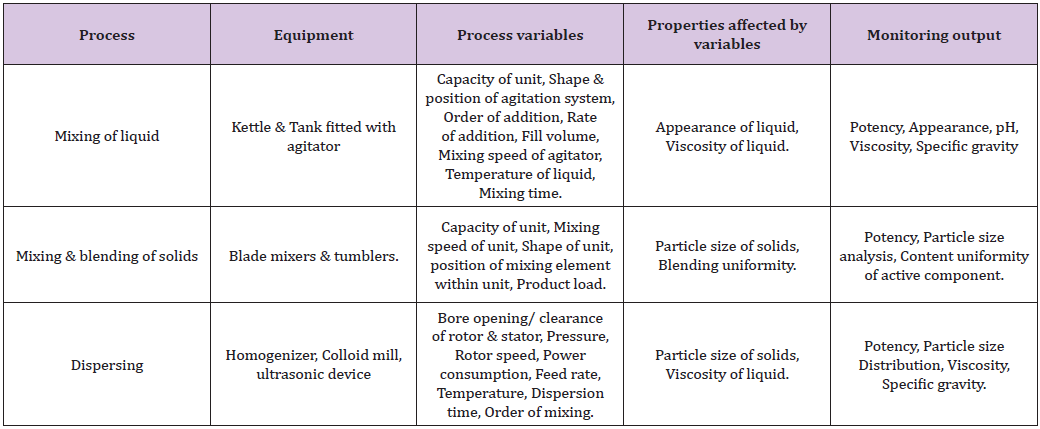
There are five unit operations in manufacturing of semisolid dosages form.
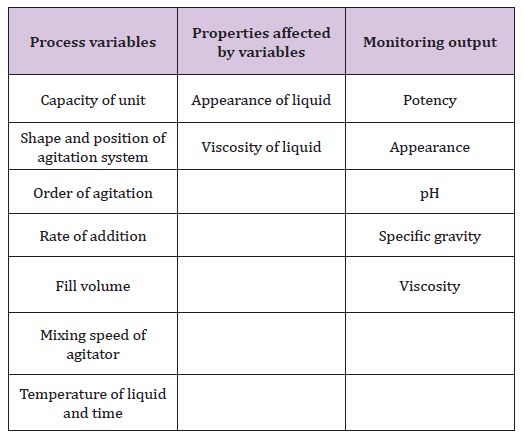
I. Mixing of liquid (Table 3)
Table 3: Process variables, properties affected by variables and monitoring output of mixing of liquids.
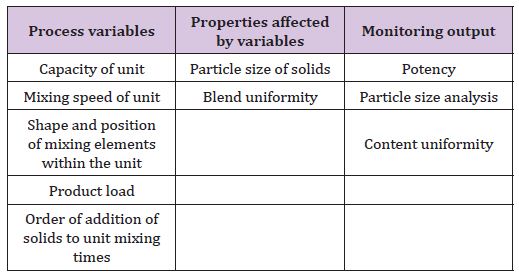
II. Mixing of solid (Table 4)
Table 4: Process variables, properties affected by variables and monitoring output of mixing and blending of solids.
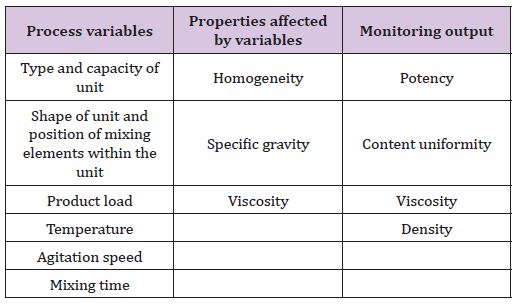
III. Mixing of semisolid (Table 5)
Table 5: Process variables, properties affected by variables and monitoring output of semisolid.
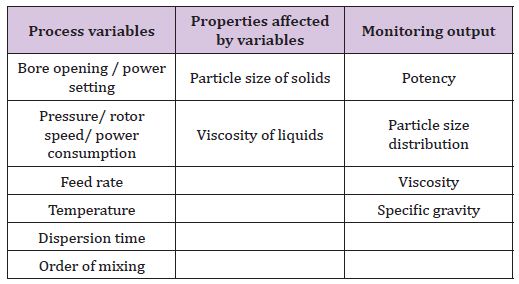
IV. Dispersing (Table 6)
Table 6: Process variables, properties affected by variables and monitoring output of dispersing.
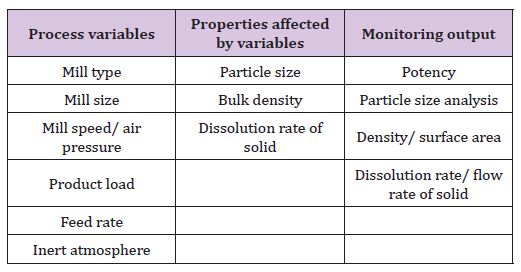
V. Milling and size reduction of solid and semisolid(Table 7)
Table 7: Process variables, properties affected by variables and monitoring output of milling and size reduction of solid and semisolid.
Validation is a proven assurance of the process efficiency and sturdiness and it is the full fledged quality control tool for the pharmaceutical industries. It eliminates the chances of batch failures as the products are manufactured as per pre optimisation of each manufacturing steps. The conventional process of testing at last stage created much problems in maintain uniformity of each batch but with the introduction of concept of validation, it has been easy to maintain the batch uniformity of the product along with imparting quality in them. This paper summarises the process validation stages of solids, liquids and semisolids which are the most common pharmaceutical dosages form in use.

























