info@biomedres.us
+1 (502) 904-2126
One Westbrook Corporate Center, Suite 300, Westchester, IL 60154, USA
Site Map
Received: October 10, 2017; Published: October 17, 2017
Corresponding author: Nisha KJ, Department of Periodontics, Vydehi Institute of Dental Sciences and Research Centre, EPIP Zone, Whitefield, Bangalore, Karnataka, India
DOI: 10.26717/BJSTR.2017.01.000447
Recent advances of proteomic meth¬odologies have opened new opportunities to obtain relevant information on normal and abnormal processes occurring in the human body. Identifying unique patterns of protein expression, or biomarkers, associated with specific diseases is one of the most promising areas of clinical proteomics. Advances in proteomic technologies have enabled comprehensive profiling of protein expression in cells, tissue, and body fluids. The use of proteins as biomarkers for periodontal disease has been the focus of researchers over the last few years. Unfortunately, many single protein biomarkers have proven to be unreliable. Developing new diagnostic tests that can simultaneously analyze the expression of multiple proteins may enable early detection, targeted preventive measures and individualized therapeutic intervention of periodontal diseases. This review discusses the main proteomic techniques and their potential applications in the field of periodontal diagnostics.
Keywords: Proteomics; Biological Markers; Periodontal Diseases; Diagnosis; Gingival Crevicular Fluid; Saliva
Abbreviations: FFF: Field Flow Fractionation; MS: Mass Spectrometry; MALDI-TOF-MS: Matrix-Assisted Laser Desorption/Ionization-Time of Flight Tandem Mass Spectrometry; LC-ESI-MS: Liquid Chromatography-electro Spray Ionization Tandem Mass Spectrometry; SELDI-TOF MS: Surface-Enhanced Laser Desorption/Ionization Time of Flight Mass Spectrometry; Tandem MS: Tandem Mass Spectrometry; IPI: International Protein Index.
Proteomics is the comprehensive study of proteins expressed in cells, tissues and fluids. It often involves the comparison of tissue samples from diseased and healthy people in order to identify proteins that are altered in disease [1]. Identifying unique patterns of protein expression, or biomarkers, associated with specific diseases is one of the most promising areas of clinical proteomics [2]. Proteomics is widely envisioned as a powerful means for biomedical research. Proteomic approach has allowed large-scale studies of protein expression in different tis¬sues and body fluids in health and disease. Recent advances of meth¬odologies in this field have opened new opportunities to obtain relevant information on normal and abnormal processes occurring in the human body. Proteins that are significantly altered in their expression, location or post translational modification in patients with a disease compared to a group of healthy individuals may represent protein targets for drug or bio-marker discovery. With the significant advances in proteomics technologies, protein biomarker discovery has become one of the central applications of proteomics. Proteomic technologies play an important role in drug discovery, diagnostics and molecular medicine because it is the link between genes, proteins and disease [3].
The word “proteome” represents the complete protein pool of an organism encoded by the genome. Proteins are fairly large molecules made up of strings of amino acids linked like a chain. While there are only twenty amino acids, they combine in different ways to form thousands of proteins, each with a unique, genetically defined sequence that determines the protein’s specific shape and function. In addition, each protein can undergo a variety of posttranslational modifications that further influence its shape and function. The human body may contain more than two million different proteins, each having different functions. Proteins are the main components of the physiological pathways of the cells. They serve many vital functions in the body such as catalysing various biochemical reactions (e.g. enzymes), acting as messengers (e.g. neurotransmitters), acting as control elements that regulate cell reproduction, influencing growth and development of various tissues, transporting oxygen in the blood (e.g. haemoglobin) and defending the body against disease (e.g. antibodies).
Proteomics is a relatively new area of research which deals with the inventory of proteins, the principal constituents of theprotoplasm of all cells. In a broader sense, the term ‘proteomics’ refer to cataloguing proteins of a biological subject and the monitoring of reversible post-translational modification of proteins by specific enzymes, i.e. phosphorylation, glycosylation, acylation, phrenylation, sulfurization, etc. In many ways, proteomics is similar to genomics. Genomics starts with the gene and makes inferences about its products which are proteins, whereas proteomics begins with the functionally modified protein and works back to the gene responsible for its production. Proteomics helps in understanding of alteration in protein expression during different stages of life cycle or under stress condition. Also, proteomics helps in understanding the structure and function of different proteins as well as protein-protein interactions of an organism. In general, proteomic approaches can be used
i. For proteome profiling,
ii. For comparative expression analysis of two or more protein samples,
iii. For the localization and identification of posttranslational modifications,
iv. For the study of protein–protein interactions [4].
A minor defect in protein structure, its function or alternation in expression pattern can be easily detected using proteomics studies. This is very important with regard to understanding of various biological processes occurring in the body.
A flow chart describing the basic steps involved in proteome analysis is given in figure 1.
Figure 1 : Flow chart showing the basic steps involved in proteome analysis.
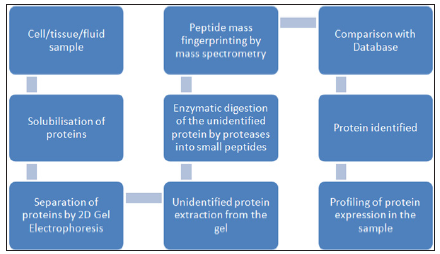
Steps commonly involved in proteomic analysis are:
i. Protein extraction from the sample
ii. Separation of proteins
iii. Peptide fractionation and identification
iv. Storage, manipulation, and comparison of the data using bioinformatics.
This step involves extraction of protein samples from whole cell, tissue or sub cellular organelles followed by purification using density gradient centrifugation or chromatographic techniques. Many components of the sample may interfere with analysis. Insoluble substances can be removed by centrifugation. The salts can be removed prior to analysis by dialysis, protein precipitation or reverse-phase chromatography. Complex samples should be fractionated before analysis to obtain simpler sub fractions. The prepared sample is then subjected to 2DE (Two-dimensional gel electrophoresis).
Two-dimensional gel electrophoresis is applied for separation of proteins on the basis of their iso electric points in one dimension and molecular weight on the other. Spots are detected using fluorescent dyes or radioactive probes. It is a powerful separation technique, which allows simultaneous resolution of thousands of proteins. The 2D gels are digitized and the resulting gel images are quantitatively and qualitatively analyzed using specialized software programs [5,6]. Although 2D-GE is a powerful technique, one of its limitations is that 2D gels remain relatively low throughput and require large amounts of starting material (~50μg) with low sensitivity for detection of low abundance proteins such as cytokines and signalling molecules. In addition, certain basic proteins, and very high- or very low-molecular weight proteins are not separated well by 2D-GE [7]. Another technique is Field Flow Fractionation (FFF) which separates proteins based on their mobility in presence of an applied field, such as electrical, gravitational, centrifugal etc [8].
The separated protein spots on gel are excised and digested in gel by a protease. The most commonly used enzyme for protein digestion is trypsin. The eluted peptides are identified using mass spectrometry.
The use of mass spectrometry (MS) to identify and characterize biological molecules is a fundamental technology in protein biochemistry and proteomic analysis. The strategies used to prepare proteins or more complex proteomic samples for MS analysis involve many steps, and in bottom-up proteomics, the protein is first broken up into peptides, either by chemical or enzymatic digestion, prior to MS analysis by either matrix-assisted laser desorption/ ionization-time of flight tandem mass spectrometry (MALDI-TOFMS), liquid chromatography-electro spray ionization tandem mass spectrometry (LC-ESI-MS) or surface-enhanced laser desorption/ ionization time of flight mass spectrometry (SELDI-TOF MS) [9-11]. The process of protein identification through mass spectrometry is done in two main ways:
i. Peptide Mass Fingerprinting
ii. Tandem Mass Spectrometry
Peptide mass fingerprinting typically uses the masses of peptides derived from a spectrum as to check against a database of predicted peptide masses. These predicted masses are recorded from digestion of a list of well documented proteins. Determined amino acid sequence is finally compared with available database to validate the proteins [12].
Tandem Mass Spectrometry (Tandem MS) utilizes collisioninduced dissociation. This process breaks proteins within the peptide backbone and because of this fragmentation, comparisons between the observed fragment sizes and the database of predicted masses is possible. A very similar method to the Tandem MS approach is peptide fragmentation fingerprinting or ‘PFF’. Instead of utilizing collision-induced dissociation, this method uses enzymatic digestion of a single peptide to generate a fragmentation pattern. These fragments are analysed and compared to a database of observed fragments for the particular enzyme in a method similar to the Tandem MS approach. Tandem MS and peptide fragmentation fingerprinting can also be used for protein sequencing [13,14].
The data obtained from mass spectrometry must be interpreted against protein databases. Sequences are usually determined using one or more search algorithms linked to an appropriate database, such as the International Protein Index (IPI) at the European Bioinformatics Institute or the nr (non-redundant) protein sequence database provided by NCBI. Several software packages are available for the analysis of mass spectrometry data. Commonly used search algorithms include SEQUEST, X! Tandem, and MASCOT [15].
Protein detection using highly sensitive proteomic technologies has allowed researchers to accurately monitor changes in biomarker proteins that are representative of disease. Proteomic analysis examines thousands of proteins at one time allowing the detection of specific protein patterns expressed as a consequence of abnormal cellular function or cellular interactions. Proteomic technologies will play an important role in drug discovery, diagnostics and molecular medicine because it is the link between genes, proteins and disease. Advances in proteomics may help scientists eventually create medications that are personalized for different individuals to be more effective and have fewer side effects.
Analysis of human body fluid proteome has become one of the most promising approaches to discovery of biomarkers for human diseases. Oral fluid diagnostics, more specifically salivary diagnostics has become one of the widely researched areas in recent years. Protein biomarkers for oral cancer, Sjogren’s syndrome and breast cancer have been recently identified in saliva [16-20]. Also, efforts are being made to apply salivary proteomics for diseasespecific biomarker discovery such as lung, gastric, uterine and pancreatic cancer [21]. Proteomic studies conducted on human saliva using a combination of liquid chromatography, electro spray tandem mass spectrometry and 2D-electrophoresis have led to the identification of 309 separate proteins [22]. Alteration of proteome levels in saliva and GCF with periodontitis has been documented and the identification of these markers could be used as a potential diagnostic tool for periodontal diseases [23].
Clinical and radiographic assessment of periodontal disease remains the basis for patient evaluation. These diagnostic measures for periodontal disease provide information primarily about disease severity and are not useful measures of disease activity. The diagnosis of active phases of periodontal diseases and the identification of patients at risk for active disease represents a challenge for both clinicians and clinical investigators. Under diagnoses within general practice leads to relatively low rates of therapeutic intervention and significant amounts of untreated disease. This emphasizes the need for the development of new diagnostic tests that can detect the presence of active disease, predict future disease progression, and evaluate the response to periodontal therapy, thereby improving the clinical management of periodontal patients [24].
<For the past two to three decades, researchers are focusing on the utility of individual biomarkers of periodontal disease activity, measured within various sources like saliva, serum, sub-gingival plaque, tissue biopsies and gingival crevicular fluid. As often happens when new technology is introduced, there are many expectations and hopes. There is no exception for clinical proteomics, however, major challenges still remain. One major challenge is to optimize the detection of low abundance proteins (cytokines, transcription factors, and cell-signalling proteins) in tissue, plasma, and body fluids, which are found in the nanogram and picogram range. Moreover, further improvement in software development for data acquisition and interpretation is needed. A good understanding of data management, correlation, interpretation, and validation is crucial to obtain accurate and meaningful results.
Based on the information presented, it is clear that the application of proteomics to the medical field has a great potential for the improvement of diagnostics and therapeutics. Nevertheless, there are challenges that need to be overcome. It is necessary to have a good understanding of the variability sources that may contribute to error such as pre-analytical, analytical, and biological variation. Pre-analytical variability may be introduced during specimen collection and manipulation, pipetting, and dilution of samples. Careful consideration should be given to specimen collection using different tube type, coagulation times, and storage conditions. Analytical variability may occur in inaccurate calibration of instruments (MS/2-DE), standardization of output, and appropriate controls and proper bioinformatics methodology. In addition, it is important to account the biological variability due to gender, age, race, and fluctuations that may occur daily within an individual (biorhythm, fasting, time of the day). All these variables may induce changes in that are not pathological in nature but that have to be differentiated from a pathological-induced process [25].
Proteomic and microbial studies have been used to successfully identify biomarkers for periodontal disease. The development of the employed proteomic platform technologies will help complete in breadth and in depth the protein profiles of periodontal disease [26]. Choi et al. [27] searched for potential protein biomarkers for periodontitis in gingival crevicular fluid using LC tandem MS (LC-MS-MS). Azurocidin, an antibiotic protein of azurophil granules with chemo tactic activity, was identified as up regulated in gingival crevicular fluid. ELISA was then used to verify upregulation of azurocidin, identifying the latter as a candidate biomarker for inflammatory periodontal disease [27]. Despite the promising prospects of using whole saliva for screening for periodontal diseases, most proteomic studies have been confined to 2-D-gel electrophoresis approaches that only used MALDI and electrospraymass spectrometry for identification of protein spots [28-31]. Only few studies have used the highly sensitive gel free LCMS/ MS approaches for studying periodontitis-related changes in the composition of whole saliva GCF, and periodontal tissues [32- 35]. The most frequently detected proteins in these studies were actins, keratins, histones, annexins, protein S100-A9, HSPB1, LEG7 and 14-3-3, apolipo protein A-I, ALB protein, albumin and serum albumin.
Gesell Salazar in 2013 identified characteristic protein patterns that differ in whole saliva of periodontal diseased and periodontal healthy subjects using a label free quantitative proteome approach analysis. In total, 20 proteins showed 1.5-fold difference in abundance between controls and patients (p < 0.05); the majority of these proteins showed higher abundance in the periodontal diseased subjects. Functional annotation of proteins linked the periodontal diseased status with acute phase response and inflammatory processes. The strongest differences were observed for protein S100-P (fold change 2.4), plastin-2 (fold change 2.2) and neutrophil defens in (fold change 2.1). They concluded that label free proteomic analysis of whole saliva is a powerful tool to characterize the periodontal disease status and differentiate between healthy and periodontal disease subjects (Figure 1).
Recent advancements in mass spectrometric proteomics provide a promising result in utilizing oral fluids to explore biomarkers for diagnostic purposes. Clinical proteomics offers the promise of biomarker discovery and early detection, diagnosis and prognosis of disease, but major challenges still remain. Further advances in technology are needed to eliminate proteomics deficiencies and augment its contributions to the medical and dental field.

























