info@biomedres.us
+1 (502) 904-2126
One Westbrook Corporate Center, Suite 300, Westchester, IL 60154, USA
Site Map
Received: September 15, 2017; Published: September 26, 2017
Corresponding author: Jean Claude Perez, £7 avenue de Terre-rouge F33127 Martignas Bordeaux metropole, France
DOI: 10.26717/BJSTR.2017.01.000394
Fundamental research published enables us to unify, overall, the genomics and proteomics information of any genetic sequence. This fundamental law is based on a bio-mathematics unification of all genetic information (bio-atoms, nucleotides, codons, amino acids, DNA/RNA/ Proteins) from the level of the six basic bio-atoms C O N H S P to the global level of whole genomes. Concretely, the dynamics of this Genomics/ Proteomics Coupling is appeared as two correlated curves translating topology and the dynamic evolution of a hierarchical classification of codons throughout the studied sequence. More precisely, the study of these curves shows that this tool highlights the functional areas and active sites within proteins. The specific context of Proteins Self-Assembly area could be an interesting field to run and improve our new basic research approach. Particularly, studying Prions by this method we propose a possible Prion PRION self-interaction with a Genomics/ Proteomics correlation coupling ratio of r=99.3 Such prospects oblige us to propose this new theoretical biomathematics approach in the study of the Genetics of the disease of Alzheimer, particularly at the level of Amyloid Plaques.
Keywords: Biomathematics; Master Code; DNA; Alzheimer diseases; Amyloid Plaques; Genomics; Proteomics
For more than 25 years, we have been looking for biomathematical laws controlling and structuring DNA, genes, proteins, chromosomes and genomes [1,2]. In 1997 we discovered a simple numerical law based solely on atomic masses, unifying the 3 languages of biology: DNA, RNA, and amino acids. This law, “the Master Code of Biology” is published in 2009 in the book Codex Biogenesis [3] and then in 2015 in a reference article peer review [4]. In 2017 we publish different applications: HIV, SNPs, brain genetics [5-8]. On the other hand, at the beginning of the 2000s we studied the integrity of the Prions proteins showing common invariant bi-attractors in “W”, while the Amyloid was characterized by a pattern in “V” shape.
Since that time, biologists such as Claudio Soto have visually demonstrated in 3D the same biological form in horseshoe for the amyloid protein [9-12]. One suspects the APP (Amyloid Precursor Protein) of playing a central part in the disease of Alzheimer. However, this role would not come from the whole gene (long of 770 amino acids), but rather of a small peptide which is detached some by “cleavage” (A-beta1-42), length from 41 to 43 amino acids, it accumulates to form the “amyloid plaques “, one lends also a neurotoxic role to this small peptide.
The therapeutic hopes are multiple their backgrounds are based on a possible blocking of the “complex chains cleavage “, while neutralizing, for example, the enzymes beta-secretase and gamma-secretase, which starts to be identified. Another promising way consists also and especially to understand the causes of the Accumulation and the Agregation of these small peptides leading to the formation of Amyloids Plaques, causes associated with the Disease At beginning of years 2000, we obtain results new and not yet published in the three principal tracks of research which are the genes TAU, ApoE4 and BetaA4 Amyloid In the research way of Professor Avila (Madrid), we are studying the TAU gene [13] by our technology reveals strong Genomics/ Proteomics coupling ratios (r=80%). In other hand, we propose a theoretical Unification of the four splicing variants transcripts of the TAU gene (Epitopes 1, 2, 3 and 4). Then, the common consensus area within TAU is, for all four variants, the first TAU/MAP repeat MOTIF…! One knows that this patterned motif constitutes, very precisely, the “tubulin-binding motif”, constituying a major key element of the functional role of TAU in Alzheimer disease.
We propose also possible interaction Simulations-based perspectives involving possible binding/assemblies between the three major Alzheimer genes: APP, TAU and Apoe4. These possible interactions (at a theoretical simulation level) correspond to strong couplings Genomics/Proteomics of the assemblies thus formed. Particularly, we propose a self-assembly TAU TAU with a very higth level coupling (r=94.2%).
Lastly, concerning the formation of the amyloid plaques, we studied possible self-interactions between peptides Beta A4 1-43 and A Beta 42: We propose a possible interaction of Amyloid with its self reverse! The figure below shows this strong link. One measures visually the strong Genomis/Proteomis. Coupling correlation of this Amyloids assembly. Moreover, our simulationbased thesis is encouraged by the recent results of Doctor Claudio Soto (Sereno Genova) which bring to us the proof of a symmetrical conformation of Amyloid peptide. There is this symmetry which would allow it “mirror-like reverse self-interaction “that we will suggest. In addition, our results are correlated with the published pathogenicity increases associated with certain mutations of Amyloid peptide Abeta42 (changes A21G, E22Q, E22G and E22K).
We discussed these results with Prion Protein discoverer, the American Nobel Prize winner Stanley Prusiner [9,12].
In does the study of the APP, our discovery make it possible to highlight certain functions? In particular, does it underline the area of the “cleavage” making emerge small peptide (and perhaps functional causes of this cleavage)? Lastly, that to conclude from the analysis of the small peptide according to this technique? And, especially, as we in addition propose it for Prion, could our simulation tool be able to propose possible scenarios of selfassembly between these small peptides likely to lead to the formation of famous “Plaques Amyloids “?
At the time of the study of gene APP, it appears three types of remarkable results:
A Bad “Genomics/Proteomics Coupling” Of Gene App: Contrary to the majority of the sequences studied up to now, where the genetic agreement reveals linear coefficients of correlation genomics/proteomics always higher than r=0.90 (90%), here, on the contrary, the genetic agreements between genome and proteome of the APP are weak (the maximum obtained is r=0.49). That means, most probably, that this protein is Heterogeneous and not very stable in its native form.
Conditions Of Emergence Of The Small Peptide By “ Cleavage “: The small peptide of 41/43 amino acids of sequence:
a) DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIAT
Is localised towards the end of gene APP (between bases 2013 to 2142, codons 671 to 714).
A study focused on this area provides three good reasons which could justify that this small peptide separates, by “cleavage” of the protein precursor APP:
1. This area (towards codons 660 more particularly) appears as very functional as of the analysis of the whole gene APP, length of 770 codons.
2. Contrary to the other areas of APP, this small area has a very good genomics/proteomics coupling ratio (higher than r=0.9), as of the general study of complete gene APP.
3. In the neighbouring areas of the address of peptide (addresses located upstream and downstream), it very quickly appears very important increasing dissensions and constraints between genomics curve and proteomics curve. These regions of dissensions can be interpreted like areas of potential Cleavages.
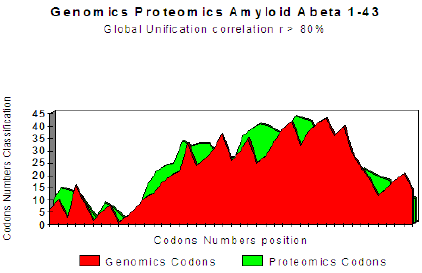
When small peptide is analyzed alone, it appears, on the contrary, an excellent Agreement Genomics/Proteomics Coupling. One measures a linear correlation “r” higher than 0.8 (80%). Moreover, in this analysis, the amino acids 9/10 would be very functional. We will note that the Biologists observe this peptide under varied lengths. However the pathogenic forms seem to correspond to lengths 43, and 42 more particularly. Let us study coupling Genomics/Proteomics in the case of the peptide of 43 amino acids, often called ” betaA4 1-43” or also ”Abeta 1-43 ”.
The two curves below show excel it coupling Genomics/ Proteomics, whose linear coefficient of correlation “r” is higher than .801. One clearly notices an optimal site towards codon 10 (Figure 1).
Figure 1: Master Code Genomics/Proteomics matching for Amyloid peptide.
Our strategy consisted in seeking possible configurations of interactions peptide-peptide. Such a discovery would constitute a beginning of track towards the understanding of the formation of “Amyloids Plaques “We validated such a scenario in the interaction between two Prions why not also between two Amyloids related to the disease of Alzheimer. Remainder, certain authors note disconcerting analogies between Prion and Amyloid:
a) In both cases, the BETA helixes are in the central part of the interaction process then contribute to the Pathogenicity.
b) Researchers observed an analogy between these BETA areas of Peptide AMYLOID in its area 15-37 (KLVFFAEDVGSNGAIIGLMVGG) and another region within Prion in 120-142 (KHMAGAAAAGAVVGGLGGYMLGSAM).
Figure 2: Amyloid beta A4 reverses self-assembly.
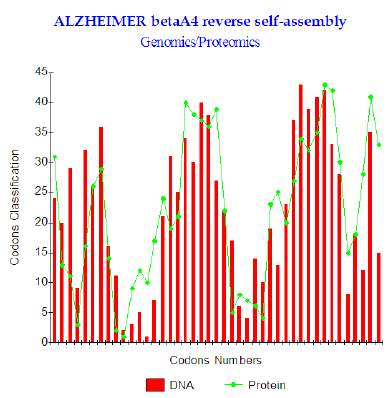
Finally, after having imagined and simulated various assumptions of interactions (shifts, role of the active site codon 10 etc…) It appeared that one of the scenarios functions too well: It consists in placing in opposite peptide Abeta 1-43 and its turned over homologue “ head-digs” After research of the OPTIMAL related position there appears that the optimal interaction puts facing Abeta 1-43 and its reverse shifted by one codon, therefore interfering, starting from the second codon (Figure 2). The levels of couplings GENOMICS/PROTEOMICS are very important: the maximum reached is r = 77.5% (Figure 2). The configuration of two put peptides face-to-face discussion is then:
1) Abeta 1-43: DAEFRHDSGY EVHHQKLVFF AEDVGSNKGA IIGLMVGGVV IAT
2) Reverse shift1:TAIVVGGVM LGIIAGKNSG VDEAFFVLKQ HHVEYGSDHR FEA
We even simulated the case, hypothetical, of a possible peptide of 42 amino acids corresponding to codons 2 to 43 and interfering is about r = 80.6%. Here this example (theoretical) (Figure 3). In fact, this peptide of 42 amino acids is very strongly associated with the experimental pathogenic situations, but it is the Abeta42 referenced peptide, obtained by cleavage of the section of integrating APP codons 1 to 42 included. What does it occur in such a case? And, more particularly, than to say certain changes which were associated with the Pathogenicity of this peptide…?
Figure 3: Amyloid A beta 42 Master Code Genomics/ Proteomics matching.
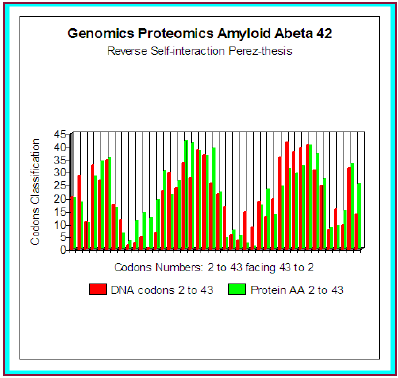
Now, the configuration studied now is thus:
1) Wild-type: DAEFRHDSGY EVHHQKLVFF AEDVGSNKGA IIGLMVGGVV IA
2) Reverse : AIVVGGVMLG IIAGKNSGVD EAFFVLKQHH VEYGSDHRFE AD
Table 1: Master Code Genomics/Proteomics % in the cases of pathogenic Amyloids.
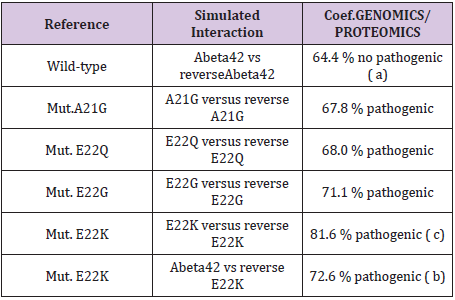
We simulated six configurations that are certain changes of which it was established than there are Pathogenes. In all do these cases; we test our thesis of “reverse-assembly” Is there a correlation between our simulated forecast and the experiments published? (Table 1) (Figures 4-6). One observes well here how the changes of the amino acids 21 and especially 22 increase coupling Genomics/ Proteomics of our scenario… knowing that they correspond, Therefore, with an increase in Pathogenity. We will point out that these two points of significant mutations are just at the median point of peptide (then, also, in the middle of its reverse one!). Effectively: 21=42/2…
Figure 4: Genomics/Proteomics % in the case of non pathogenic wild type Amyloid (a).
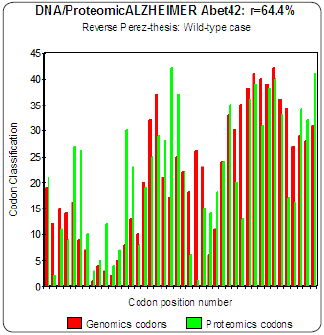
Figure 5: Genomics/Proteomics % in the case of pathogenic Mut E22K Amyloid (b).
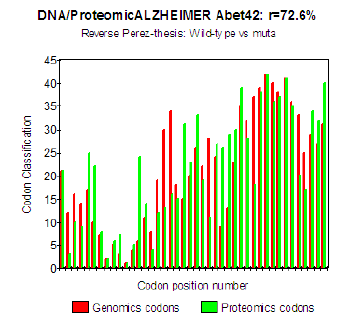
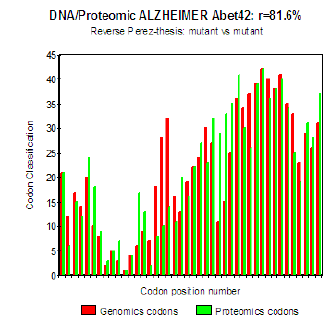
Figure 6 : Genomics/Proteomics % in the case of pathogenic Mut E22K Amyloid (c).
The three curves above correspond to the three noted cases (a), (b) and (c) in the preceding table.
There are interested more particularly in the case of mutant E22K, there make it possible, by comparison, to locate the causes of this increase in coupling when one passes from the case of interaction between two
(a) Wild-types to the hybrid case of interaction of a mutant with a wild-type
(b) Then, finally, to the case of interaction of two mutants together
(c) One observes also the attenuation of the peaks of differences Genomics/Proteomics in all first half of the curves (codons 1 to 20: attenuation of the variations green/red) what translates an improvement of the Agreement of Genomics/ Proteomics Coupling and, also, of the Pathogenicity!
We here have just shown how:
a. In a major therapeutic field, the Alzheimer disease,
b. A completely new theoretical tool, “Master Code of Biology“.
c. Those of a law of Unification Proteomics/Genomics,
d. Allowed to imagine, discover, test and simulate a scenario of “self-assembly “ between Amyloids peptides which could explain the formation of “Amyloids Plaques “, central symptom associated with the disease of Alzheimer.
e. Lastly, our thesis is seen reinforced by simulations of case of known mutations points which one knows associated with the Pathogenicity.
All these data illustrate how a new fundamental discovery could very quickly be applied and used by the research world scientific community.
As demonstrated by Pr claudio Soto in [10] “Alzheimer’s disease transmission may be similar to infectious prion diseases”. Then particularly the Master Code Of Biology biomathematical approach reveals also strong relationships between our simulations on Prions and on Amyloids self-assemblies. Both Prion and most probably Amyloid are proteins whose functionings are beyond the state of the conventional art of Biology. Therefore, we believe that multidisciplinary approaches close to quantum physics may allow us to understand how they work [14].
We thank several researchers interested in our approach to the « Master Code of Biology »: Professor Luc Montagnier (Paris), Professor Claudio Martinez Debat (Montevideo), Dr Volkmar Weiss (Leipzig), Professor Diego Rapoport (Buenos Aires), Dr Marco Francisco Paya Torres (MD Alicante), Gilles Roquette (Lille), Perry Marshal (Chicago), Dr Robert Friedman (MD Santa Fe), Dr François Gros (Pasteur Institute, co-discoverer of RNA messenger with James Watson and Walter Gilbert). Thank Professor Stanley Prusiner (UCSF https://ind.ucsf.edu/ind/aboutus/faculty/prusiners) and Professor Jesus Avila (Madrid) for various discussions on this research.

























