info@biomedres.us
+1 (502) 904-2126
One Westbrook Corporate Center, Suite 300, Westchester, IL 60154, USA
Site Map
Received: July 20, 2017; Published: August 01, 2017
Corresponding author: Hassan M Yaish, Department of Pediatrics, University of Utah, school of Medicine, Salt Lake City, UT 84113, USA
DOI: 10.26717/BJSTR.2017.01.000242
Keywords:VWD type 3; Inhibitors; re-FVIII; Bethesda units
Abbreviations: VWD: Von Willebrand Disease; PD: Plasma Derived; PT: Physiotherapy; CFC: Clotting Factor Concentrate; BI: Bolus Infusion; CI: Continuous Infusion
Type 3 VWD is the least common and most severe among the three types of VWD. The incidence was estimated at 5-3/million and the severity is thought to be similar to mild or moderate hemophilia A. A recent report from the UK showed that failing to perform all required assays for the diagnosis of VWD Type 3, may occasionally result in giving a wrong diagnosis such as mild or moderate hemophilia A, a practice occasionally used by some hemostasis laboratories.[1] Type 3 VWD is usually associated with a clinical course that combines the mucous membranes bleeding features seen in patients with VWD as well as the deep muscle and joint bleeding encountered in patients with mild to moderate hemophilia. The lack of VWF, which usually protects and delays the degradation of FVIII result in very low level of FVIII activity, which contributes to the bleeding severity. The treatment of this condition is somewhat similar to that of patients with hemophilia which consists of infusions to replace the missing factors as on demand regimen using plasma derived (PD) products which contains both FVIII and VWF. Furthermore, many of the patients are currently on some form of prophylaxis to eliminate or decrease the frequency of bleeding episodes.
7.5-9.5 % of the patients develop inhibitors to VWF becoming non- responsive to replacement therapy, and prone to develop severe anaphylactic and life threatening reactions when exposed to any product that contains VWF [2]. A previous exposure to VWF , extent of the exposure, as well as mutations causing a null-allele or deletion of the gene are all known predisposing factors for the development of inhibitors in patients with type 3 VWD. Clinical course variability in type 3 VWD is attributed to the different antigenic regions on the gene and the potency of the inhibitor. Bleeding manifestations could be very severe in some patients presenting with long refractory and recurrent epistaxis, GI bleed, joint bleeds or formation of pseudo tumor in various tissues. It is somewhat surprising that very few reports on inhibitors in type 3 VWD were published in the USA in comparison to the European literature. The neutralizing antibodies are usually polyclonal IgG class which precipitates VWF in normal plasma without any effect on purified FVIII unless it is bound to VWF, and for this reason, recombinant FVIII is usually effective in controlling the bleeds , keeping in mind that its half-life in such patients is very short at approximately 3 hours [3,4].
To describe the clinical course in patients with type 3 VWD who develop inhibitor to the factor, and the circumstances leading to the diagnosis of neutralizing antibodies in type 3 VWD, the diagnostic tests required, and the treatment options.
We report here on two young women with type 3 VWD, the first was diagnosed at birth after prolonged bleeding from a heel stick, and the second at the age of 4 years. Both had significant nose bleeds, traumatic joint bleeds bruises and hematomas at the site of injections. Both have frequent and prolonged exposures to PD products containing FVIII and VWF and both have developed inhibitor to VWF but ran two completely different clinical courses, so they will be presented separately. In the first patient, severe anaphylactic shock developed once she is infused with any of the various PD products available at the time, suggesting that the reaction was not due to intolerance or allergy to one certain product [5].
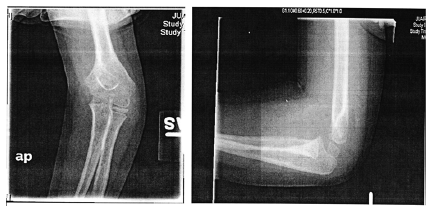
Figure 1: Left Elbow Arthropathy.
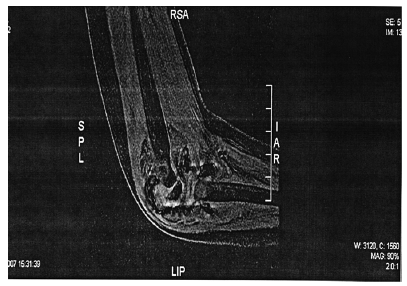
Figure 2: Chronic Elbow Arthroplasty.
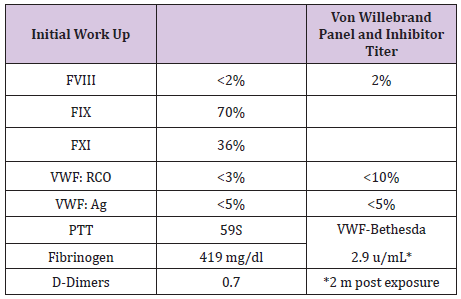
Table 1: B.J Initial Workup and PTT Correction.
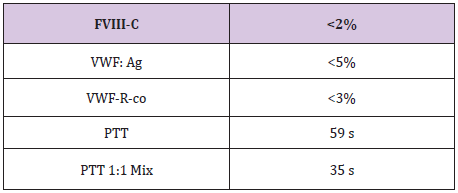
Table 2
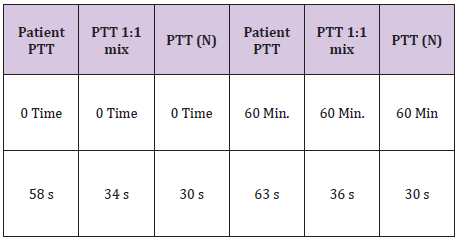
Table 3: Lab work for mixing study.

Table 4: VWF- RCF.
BETHESDA Quantitative 2.9 u/mL*
The second patient became refractory to factor replacement and failed to respond to the PD products, she continued to bleed and failed to raise her FVIII or VWF after repeated infusions. Two months after last exposure both girls were tested for the presence of alloantibody to VWF with a Bethesda assay using ristocitin cofactor, which revealed low titer inhibitor in both.
B.J a neonate, who bled from a heel stick site, had bloody tracheal aspirate and stool and found to have VWD type 3. Tables 1 & 2 At 2 years of age she injured her Left elbow, developed a bleed in the joint and supracondylar fracture (Figure 1). An MRI obtained later on showed hemophilic arthroplasty (Figure 2) Physiotherapy (PT) was planned after Clotting Factor Concentrate (CFC) infusions. A life threatening reaction with hypotension, wheezing and tongue edema developed after her fifth dose of Humate-P. Not knowing the nature of the reaction, both Humate-P as well as Alphanate infusions were attempted again after heavy premedication, but resulted in similar anaphylactic reactions. Lab work for mixing study (Table 3) and Bethesda assay, (Table 4) revealed the presence of alloantibody; presence of alloantibody to VWF rendering her resistant to the PD products and requiring an alternative treatment for bleeding episodes. r-FVIII was successfully used in treating her future bleeds. VWD Type3/Patient #2
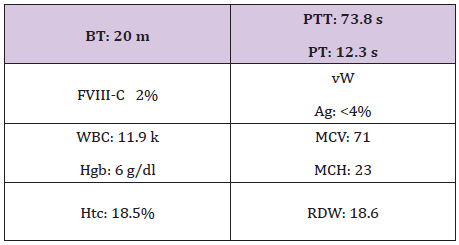
Table 5: VWD type 3.
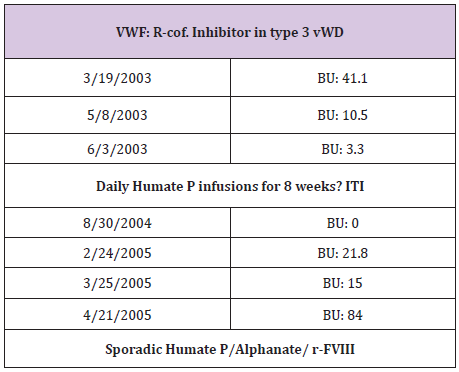
Table 6: Lab tests revealed alloanti bodies to VWF.
R.W is the second patient who was seen at 4 years of age when in- patient for severe, refractory life threating nose bleed requiring intensive CFC infusions, surgical intervention, and blood transfusion. She was also diagnosed with VWD type 3 (Table 5) and received multiple infusions for subsequent bleeds. At the age of 6 years, she showed poor response to both Humate-P and Alphanate in treating a severe nosebleed. Lab tests revealed alloanti bodies to VWF (Table 6), and was treated with continuous infusions re- FVIII. ITI with daily Humate-P was attempted and continued for 4 months with significant drop in the titer (Table 6) and no significant anaphylactic reactions. R-FVIII administered by bolus Infusion (BI) or continuous infusion (CI) was effective in traumatic hip bleed as well as for appendectomy. Menorrhagia was well controlled by hormonal therapy and occasional r-FVIII infusions, but most recently monthly prophylaxis with r-FVIII infusions during menstrual periods was initiated to prevent or minimize excessive bleeding [7].
Severe anaphylactic reactions are frequently seen in somepatients with type 3 VWD who develop alloantibodies to VWF. Alloantibody in this setting did not increase the severity of bleeding manifestation. Spontaneous joint bleeding was not encountered since the bleed in both patients was traumatic. r-FVIII given as BI at short intervals, or as CI, was proved effective in treating bleeds. ITI is feasible in some patients, however, the risk of anaphylactic reaction, lack of evidence of efficacy, and the availability of alternative therapy, have all contributed to lack of popularity of such approach.
Based on several reports in the literature, the mutations in our patients are expected to be that of large deletions or null-allele in the VW gene. In the first patient, a frame shift alteration in exon 38 caused a novel homozygous or hemizygous insertion was described (6749_6750 ins C; Pro 2250 fs). In the second patient, a partial DNA testing revealed the absence of most of the previously reported large deletions, so far leaving a high possibility that a mutation that was not described before will be identified.
Alloantibody in type 3 VWD is a rare event of a rare disease. Only few reported cases in the US literature in contrast to Europe. The condition may go unrecognized until life-threatening reactions occur upon re-exposure to VWF, or antibodies are detected by screening. Delaying the diagnosis of alloantibody to VWF in patients with type 3 VWD, will eventually delay effective treatment for bleeding episodes and may expose the patient to life threatening anaphylactic reactions upon exposure. This could be avoided if regular antibody screen is considered.

























