info@biomedres.us
+1 (502) 904-2126
One Westbrook Corporate Center, Suite 300, Westchester, IL 60154, USA
Site Map
Received: July 18, 2017; Published: August 01, 2017
Corresponding author: Kriti Chauhan, Department of pathology, Maharishi Markandeshwar Institute of Medical Sciences And Research, Mullana, Ambala, Haryana-133203, India
DOI: 10.26717/BJSTR.2017.01.000232
Hemophagocytic lymphohistiocytosis (HLH) is a life threatening hyper inflammatory syndrome occurring due to ineffective immune process. It can be genetic (familial form) or secondary (acquired form). We present a case of HLH in a 50 year old male who presented with pancytopenia and huge platelet clumps on peripheral smear which subsequently leads to diagnosis after correlation with clinical, hematological and biochemical findings.
Keywords: HLH; Pancytopenia; Platelet Clumps
Abbreviations: HLH: Hemophagocytic Lympho Histiocytosis
Hemophagocytic syndrome with special mention to its secondary form is a rare syndrome which occurs after strong immunologic activation and release of cytokines characterized by unrestricted activation of T lymphocytes and macrophages invariably leading to death in absence of treatment [1,2]. It is associated with certain clinical and hematological abnormalities of which pancytopenia, hyper ferritinaemia, hepatosplenomegaly, fever and hemophagocytosis in bone marrow were observed in our case.
Figure 1: The sample was repeated to rule out EDTA effect on platelets, but this feature persisted and also showed few platelet clumps engulfing WBCs.
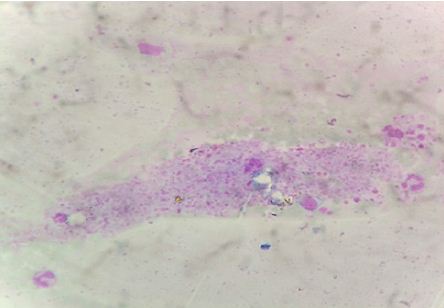
A Fifty year old male patient presented with decreased appetite since 10 to 15 days and dyspnoea on exertion associated with cough. The patient was non alcoholic and non smoker. On examination, the patient was febrile, oriented and a wheeze was heard on right lower zone. Chest X-ray showed a diffuse haziness on right lower zone. His ultrasonography of abdomen revealed hepatosplenomegaly with dilated portal vein, splenic artery and ascitis. The liver function tests were altered and revealed a high bilirubin= 3.4 mg/dl (N= 0.20 -1.0 mg/dl), direct bilirubin = 2.90 mg/dl (N= 0- 0.2 mg/dl), SGOT= 136 U/L (5-40 U/L), SGPT= 76 U/L (5-35 U/L) and Alkaline phosphatase = 621 U/L (28-112 U/L). Renal function tests and other biochemical investigations were unremarkable. Dengue serology and malaria antigen detection tests were negative. A clinical diagnosis of? Hepatitis? Lower respiratory tract infection?? Acute respiratory distress syndrome was made. The patient was non reactive for HbS antigen and HCV antibody. The hematological investigations showed pancytopenia (Hb = 6.0 g/dl, TLC= 2500/mm3 and platelets= 50,000 cu.mm). On peripheral smear examination, RBCs were predominantly microcytic hypo chromic revealing mild degree of anisopoikilocytosis. The differential leucocyte count was polymorphs - 69%, lymphocytes- 25%, monocytes-6. An interesting finding was presence of large number of platelet clumps or aggregates (Figure 1). The sample was repeated to rule out EDTA effect on platelets, but this feature persisted and also showed few platelet clumps engulfing WBCs. On account of this, a prothombin time and activated partial thromboplastin time was ordered. Both came out to be high. PT = 22 sec (N= 11-16 secs) and APTT = 36 sec (N =30-32 secs). The peripheral smear was examined again and it was observed that many of the polymorphs and monocytes revealed cytoplasmic vacuolation suggestive of some infectious etiology and subsequently blood culture report was positive for pseudomonas aeroginosa sensitive to meropenem, imipenem, amikacin, ceftriaxone, tobramycin and piperacillin. The sputum was also sent for culture and came out to be positive for candida.
Figure 2: The plasma cells were also present in significant numbers.
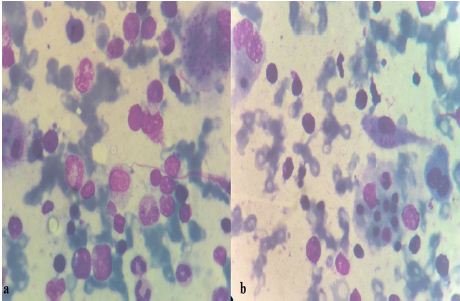
Based on all these investigations the patient was diagnosed as suffering from fungal pneumonia with hepatitis associated with portal hypertension along with sepsis and was treated for the same but on subsequent investigations, pancytopenia remained unresolved. Hence, a bone marrow examination was done. The aspirate was particulate and norm cellular for age. The myeloid and elytroid precursors were well preserved. Megakaryocytes were adequate in number. In addition to this, there was a prominence of macrophages showing phagocytosis of hematopoietic cells particularly platelets and erythroid precursors (Figure 2). The plasma cells were also present in significant numbers. A serum ferritin level was ordered which came out to be significantly high= 1650mg/dl (N=24-336mg/dl).The serum triglyceride levels were marginally high (152mg/dl) (N=40-149 mg/dl).Hence it was found that the patient fulfills five criteria of Hemophagocytic syndrome which are fever, splenomegaly & hepatomegaly with hepatitis like features, pancytopenia, hyperferritenemia and evidence of hemophagocytosis on bone marrow. The increased PT, APTT values, presence of blood clots and positive blood culture report for pseudomonas were supporting evidence of sepsis and the patient likely being in DIC. In our case this was the presumptive cause of hemophagocytic syndrome. This case is different because it was the repeated appearance of platelet aggregates on peripheral smear with pancytopenia and hepatosplenomegaly that raised the doubt of a Hemophagocytic syndrome which was further confirmed by bone marrow examination and other biochemical investigations.
Hemophagocytic syndrome also known as Hemophagocytic Lymphohistiocytosis (HLH) is a life threatening condition characterized by hyper inflammation due to immune deregulation resulting into organ damage by activated macrophages and hustiocytes [3]. HLH was first described by Scott [4] in 1939. It was subsequently divided into primary/familial form and reactive/ secondary form. The familial form has autosomal recessive inheritance and develops in children younger than 2 years. It is very lethal and may be associated with immunological diseases like Chediak Higashi and Griscelli syndromes [5]. Secondary HLH can occur at any age and may be associated with infections (EBV, Dengue, CMV, HSV, Tuberculosis, leishmaniasis, Salmonella typhi, Chlamydia etc) malignancy (ALL, Hodgkin’s lymphoma, Multiple Myeloma, Peripheral NK T Cell lymphoma etc) Collagen vascular diseases(SLE, Sjogren’s syndrome, juvenile idiopathic arthritis) [2]. HLH is a diagnosis if at least five out of following eight diagnostic criteria met.
A. Fever
B. Splenomegaly
C. Cytopenias affecting more than 2 lineages (Hb‹9 g/dl, Platelet count ‹1 lac/cumm, Neutrophils‹1000/cumm)
D. Hypertriglyceridemia≥3.0mmol/L(≥265mg/dl or hypofibrinoginaemia (less than or equal to 1.5g/L)
E. Hemophagocytosis in bone marrow ,spleen or lymph nodes
F. Low or absent NK T cell activity
G. Increased ferritin levels(≥500μg/ L)
H. Soluble CD25 (i.e soluble IL-2 receptor≥2400u/m)
Apart from these, patients frequently have altered LFT values like hyperbilirubinaemia, elevated transaminases raising possibility of hepatitis [6] and features of disseminated intravascular coagulation like Raised PT, raised APTT, low fibrinogen levels, increased D- dimer and FDP’s. [7] Our patient had altered LFT and high PT, APTT values. D- dimer and fibrinogen levels were not done.
Once a diagnosis of HPS has been established, it is essential to hunt for an underlying cause to improve life outcome. Infection associated HPS commonly occurs in immune compromised patients and is usually fatal [8] but in our case there was no evidence of compromised immunity. Shabber et al [9] have reported a 72% mortality rate in HLH with multisystem organ failure being the most common cause of death. Our patient was treated with heavy dose of antibiotics but succumbed to death. HLH and sepsis syndrome are known to overlap. NK cell activity and CD4 counts are two tests which help us differentiate these two conditions as there sensitivity is 100% in HLH.
The treatment should include a combination of chemotherapy and immunotherapy. The newest treatment protocols HLH-2004 represents a consolidation of various approaches to treatment with the motive to achieve clinical stability and then to cure with bone marrow transplantation [10]. The protocol recommends a therapy with corticosteroids, etoposide and cyclosporine A for 8 weeks with the use of intrathecal methotrexate in patients with neurological symptoms [11].
HLH is a very rare but fatal disease which may go undiagnosed and hence untreated. A strong clinical suspicion along with correlation with biochemical investigations and histopathological findings is mandatory for timely intervention. It is required on part of pathologist to carefully examine the peripheral smear and bone marrow specimens to look for any evidence of hemophagocytosis in cases of pancytopenias with hepatosplenomegaly because this can be the first clue to diagnosis of syndrome.

























