info@biomedres.us
+1 (502) 904-2126
One Westbrook Corporate Center, Suite 300, Westchester, IL 60154, USA
Site Map
Received: June 27, 2017; Published: July 06, 2017
Corresponding author: Joseph Curtis, Cascade Biotherapeutics, Inc, 4938 Hampden Ln, Bethesda, MD 20814, USA
DOI: 10.26717/BJSTR.2017.01.000173
This review presents an omics systems perspective that presents an integrated approach to understanding the contribution of the genetic, epigenetic, metabolomic, and proteomic interactions under the influence of a dynamic nutritional environment. The review takes into account the application of genomic and post-genomic era methodologies being used to study the impact of inter-individual phenotypic differences and disease susceptibility to current and future generations. Every individual alive today is a combination of the heliotypes inherited from their parents, and the results of genetic-epigenetic interaction influencing the fetal epigenome during pre-natal development and post-natal life. Epigenetic marks imprinted in-utero are attributed to deficits in the maternal contribution to the fetal nutritional environment. Integral to this discussion is the concept of developmental plasticity, which presents itself as an unfolding narrative in the “developmental origins of health and disease” (DOHaD) hypothesis, and the ‘predictive adaptive response (PAR)’ concept. When taken together, these concepts present the case for an adaptive response to developmental programming that canpredispose an individual to future diseaseor confers an advantage. Nutriepgenomics is the study of how nutrients affect human health through epigenetic modifications. Recent advances investigating how the nutritional environment during embryonic development influences the establishment of metabolic phenotypes that can contribute to the emergence of disease are discussed. Several encouraging trials suggest that prevention and therapy of age- and lifestyle-related diseases by individualized tailoring to optimal epigenetic diets including combinations of natural products, polyphenols or drugs are conceivable.
Keywords: Developmental; Diabetes; Genome; Macronutrients; Micronutrients; Metabolic; Nutriepigenomics; Nutirgenomics; Obesity; Personalized; Phenotype; Polyphenols
Abbreviations: CGD: Cytosines-Guanine Dinucleotides; DOHaD: Developmental Origins of Health and Disease; EWAS: Epigenome-Wide Association Studies; FOCM: Folate One-Carbon Metabolism; GWAS: Genome-Wide Association Studies; Hap-ASM: Haplotype-Dependent Allele- Specific Methylation; IUGR: Intrauterine Growth Restriction; LD: Linkage Disequilibrium; MQTL: Methylation Quantitative Trait Loci; miRNA: microRNA; PAR: Predictive Adaptive Response; PPL: Postprandial Lipemia; SNPs: Single Nucleotide Polymorphisms
The nutritional environment influences every stage of life. Beginning with its influence on the genetic architecture of haploid cells that participate in the creation of a new individual and continues to influence processes responsible for fetal development, cellular respiration, apoptosis, and predisposition to metabolic disease. The epigenome provides an interface between the environment and the genome [1]. Epigenetic markers are enzyme-mediated chemical modifications of DNA and of its associated chromatin proteins. These modifications play key roles in regulating genomic functions, without altering the primary DNA sequence, and are transmitted with high fidelity over many cell generations. The main epigenetic mechanisms are DNA methylation, histone modifications and microRNA, which are involved in the propagation of the chromatic structure and genome activity states [2].
Epigenetic modifications such as DNA methylation play a key role in gene regulation and disease susceptibility.DNA methylation is an epigenetic process implicated in human disease that involves methylation of cytosine, usually at cytosine-(phosphate)-guanine (CpG) dinucleotides in the promoter region or within genes. This molecular phenotype plays a pivotal role in gene expression by affecting chromatin structure and altering the availability of coding regions to transcription mechanisms [3]. In differentiated mammalian cells, DNA methylation occurs primarily at cytosines in CG dinucleotides, so called CpG methylation, which is associated with regulation of cell specific gene expression. In contrast to DNA sequence variation, epigenetic variation is sensitive to both inherited and environmental inputs [1-3].
Patterns of DNA methylation are primarily established early in life, but may also be dynamic and change in response to environmental stimulations such as diet and exercise. DNA methylation at specific loci can be influenced by sequence variations, such that individual genotypes at a given locus may result in different patterns of DNA methylation due to allele-specific methylation. These sites are called methylation quantitative trait loci (mQTLs) and can influence the methylation pattern across an extended genomic region. A distinguishing feature of mQTLs is that they are detected across ancestry, developmental stage and tissue type [4]. Concurrently, once epigenetic modifications are introduced they can be stable and inherited, making epigenetics a potentially important pathogenic mechanism in complex metabolic diseases [1-3].
A haplotype is a group of genes within an organism that was inherited together from a single parent. In addition, the term “haplotype” can also refer to the inheritance of a cluster of single nucleotide polymorphisms (SNPs), which are variations at single positions in the DNA sequence among individuals. The concept of inheritance is especially important to understanding how the nutritional environments of the parents can influence the epigenome of future generations. Haplotype-dependent allelespecific methylations (hap-ASM) can impact disease susceptibility, however maps of this phenomenon using stringent criteria in disease-relevant tissues are few in number [5]. In order to understand how hap-ASM contributes to disease, a short review is needed.
First, statistical evidence from genome-wide association studies (GWASs) has implicated numerous DNA sequence variants, mostly SNPs, as candidates for inter-individual phenotypic differences and disease susceptibility. Diseases interrogated with well-powered GWAS demonstrate that the majority of risk alleles reside in noncoding regions and have small effect sizes and would not achieve genome-wide significance in more moderately sized cohorts [2,6]. Therefore, methods to provide biological validity to both supra- and sub-threshold variants are a high priority. Moreover, multiple statistical comparisons demand stringent thresholds for significance, p < 5 × 10−8 for a GWAS 1 and this level probably leads to the rejection of many biological true positives with sub-threshold p values [4,5].
Second, a combined genetic-epigenetic approach can help to address the challenge of variant biological validity. ASM is a combination of two different phenomena: genomic imprinting, where the methylation of an allele is determined by its parent-oforigin, and (non-imprinted) hap-ASM, in which the local sequence context acts in cis to dictate the methylation status of local CpGs. Hap-ASM can be assessed either directly by bisulfite sequencing (bis-seq) in heterozygotes or by methylation quantitative trait loci (mQTL) analysis, which correlates net methylation of single CpGs with genotypes at nearby SNPs [5]. The ability to detect anmQTL is influenced by statistical power, which may depend on SNP allele frequency and linkage disequilibrium (LD) structure, which vary based on ancestry, or the variance of methylation at a specific CpG site, which may differ across stages of development or tissue type. These SNP-CpG relationships have wide-reaching biological relevance and likely contribute to inter-individual variation in gene regulation. The relationship between sequence variants and DNA methylation patterns in different sets of individuals may also provide insight into complex traits in which a particular environmental exposure associates with a trait only if incurred at a particular developmental window [4]. Mapping hap-ASM and mQTLs and superimposing these maps on GWAS data can support the biological relevance of GWAS peaks, the hypothesis being that detection of hap-ASM or an mQTL near a GWAS peak suggests the presence of a valid regulatory SNP or haplotype, which associates a physical asymmetry between the two alleles in heterozygotes [5].
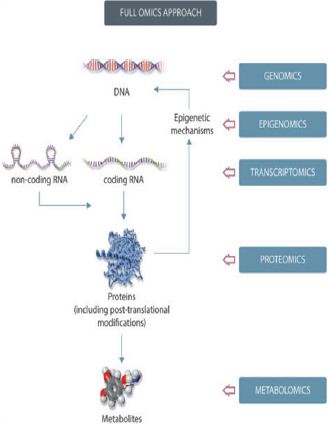
Figure 1: Schematic representation of a full omics approach to Nutrigenomics.
A major finding in the field of nutrition is discovering that dietary components may reshape the genome in utero and that epigenetic changes induced during early life may permanently alter the phenotype in the adult organism (Figure 1). A number of reviews have focused on maternal nutrition and its impact on epigenetic mechanisms along with studies addressing different types of exposure, such as nutritional factors, glucocorticoids, and endocrine-disrupting chemicals. Studies focusing on the developmental origin of health and disease have identified links between early nutrition and disease, consistent with the “developmental origins of health and disease” (DOHaD) hypothesis. This hypothesis postulates that early life development is critically sensitive to inadequate nutrition and other environmental factors, leading to permanent changes or marks in development that can influence the health of an individual in later life and increase their risk of disease [2,6,7,8].
Epigenetic modifications are stable during cell division, and they regulate gene activity states over future cell generations. During development, epigenetic marks, such as DNA methylation, histone modification and microRNA expression, undergo substantial reconfiguration, which affects the genes and pathways that are essential for both early life development and later life physiological functions. (Figure 2) Experimental studies supporting the view that prenatal nutritional environments may collaborate with the marks resulting from interaction with the parental nutritional environment contribute to induce altered metabolic phenotypes in the offspring. The combination of these interactions imparts an increased risk of non-communicable diseases, including obesity, diabetes and cardiovascular diseases to future generations of humans [1-9].
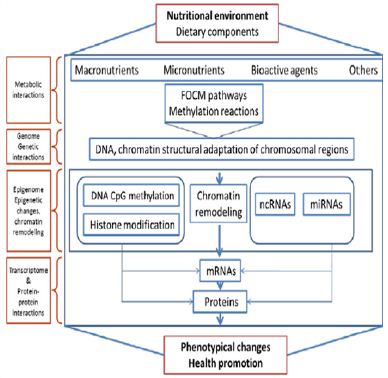
Figure 2: Phenotype Diversity: Nutritional Impacts on the Epigenome, Genome, Metabolome, Proteome and Transcriptome Contribute to Phenotype Generation and Health Outcomes.
However, insufficient knowledge exists in addressing how nutritional factors influence epigenetic mechanisms during fetal development and how to prevent potential negative effects on health. The impact of nutrition on genomic DNA methylation through one-carbon metabolism is well-documented. Specifically, it has been demonstrated that dietary deficiency or excess of the methyl group donors needed for the cellular methylation reactions can alter epigenetic patterns, which may persist for a long period and alter gene expression causing phenotypic changes [6]. The vitamins choline, folate, B6 and B12 are cofactors essential to maintaining the one-carbon transfer cycles [1,8,9].
Dietary components (amino acids, high-fat or high-glucose diet, vitamins, bioactive factors,) can affect genome function and gene expression in utero and during early life, influencing epigenetic mechanisms through folate-mediated one-carbon metabolism or transmethylation pathways to affect DNA methylation, histone, or non-coding miRNAs [6]. In contrast, the impact of diet on other epigenetic mechanisms, including histone modification, chromatin modifying proteins, and microRNA (miRNA) expression is poorly defined. Some dietary components induce favorable epigenetic effects on the organism, while others may cause unfavorable epigenetic health effects. In this respect, it is of great importance to identify both the favorable and unfavorable epigenetic impact of dietary components [8,10].
Within the DOHaD concept, an important point is that throughout embryonic and fetal development, intense epigenetic remodeling takes place that is necessary for the establishment of transcriptional programs responsible for cellular proliferation and differentiation. During these sensitive developmental periods, the epigenome is especially plastic and most sensitive to environmental disturbances. Epigenetic reprogramming during embryo development induces the most extensive changes to the epigenetic state. The epigenetic state that is set during the reprogramming stages persists at some loci for the lifetime of an individual. Evidence to support a role of epigenetics in developmental programming of disease demonstrated the impact of a suboptimal intrauterine nutritional environment on the epigenome and phenotype of the offspring [1-6].
Consequently, poor nutritional environments in early life can induce structural and functional changes in key organs responsible for nutrient regulation, including the brain, liver, adipose tissue, muscle and pancreas. Presently, this view is commonly referred to as the ‘predictive adaptive response (PAR)’ concept. Exposure to adverse environmental factors such as inadequate or unbalanced nutrient supply during in utero development may ‘program’ for the long term appetite regulation, feeding behavior, as well as adipose tissue and pancreatic beta cell dysfunction in the developing fetus. As a result of these processes, the fetus may be adapted to adverse nutritional conditions by reducing its ability to produce insulin and by occurrence of insulin resistance [1,5,11].
According to the ‘thrifty phenotype’ hypothesis, metabolic adaptation may provide a short-term survival benefit for neonates and infants experiencing a poor postnatal environment that results in an enhanced capacity to store fat in conditions of irregular availability of food resources, but may also predispose the child to T2D development in conditions of food abundance in postnatal life. Additionally, under malnourished conditions when the fetus exhibits poor growth in utero, commonly referred to as intrauterine growth restriction (IUGR), the fetal adaptation to under nutrition is realized by a variety of mechanisms responsible for the energy and glucose metabolism, such as enhanced peripheral insulin sensitivity for glucose utilization, increased hepatic glucose production, lowered insulin sensitivity for protein synthesis in muscle, and impaired pancreatic development [11].
Numerous research findings suggest that early-life adverse events including insufficient nutrition in utero might be epigenetically ‘imprinted’ and ‘remembered’ decades later, thereby permanently influencing the metabolic phenotype. There is convincing evidence that epigenetic alterations, including those triggered by early-life events and persisting through adulthood, is an important etiological factor in the development of T2D. Changes in DNA methylation and associated changes in patterns of expression of genes implicated in various aspects of glucose metabolism such as: cell dysfunction, glucose intolerance and insulin resistance, have been shown to be critically involved in the pathogenesis of T2D. The specific DNA methylation markers have been repeatedly identified in peripheral blood and pancreatic islets of the T2D patients. A schematic representation of hypothetical regulatory pathways responsible for developmental nutritional programming of T2D is presented in (Figure 3) [11].
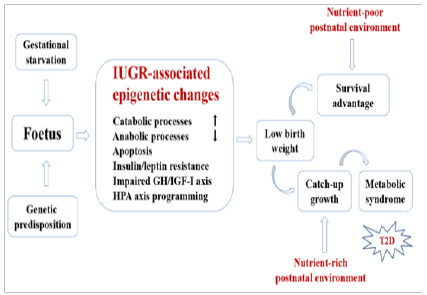
Figure 3: Schematic representation of hypothetical regulatory pathways responsible for developmental Programming of type 2 diabetes (T2D).
The long-term health consequences of the Dutch famine (‘Hunger Winter’) are the most comprehensively studied up to now. This famine was caused by the Nazi food embargo, affected the western Netherlands from November 1944 to May 1945. Such circumstances of the Hunger Winter famine provide the opportunity to thoroughly examine the link between inadequate maternal nutrition during particular trimesters of pregnancy and the offspring’s adult health status. While a normal daily ration is 2000 kcal and 2500 kcal for women and men, respectively, the average daily rations during the famine were less than 700 kcal. The prenatal exposure to the Dutch famine has been repeatedly shown to be related to the impaired metabolic phenotypes such as elevated levels of plasma lipids and body mass index (BMI), as well as enhanced risks of obesity and cardiovascular disease (CVD) later in life. Severe malnourishment during late gestation was also related to impaired glucose tolerance of the offspring into adult life [6,11-14].
In the Dutch famine study, compelling evidence has been obtained that exposure to famine during prenatal development may result in persistent epigenetic changes. Although no relationship between the prenatal exposure to the Dutch famine and overall global DNA methylation in adulthood was observed, levels of methylation of particular genes were clearly associated with prenatal famine exposure. The methylation levels of the imprinted gene encoding an insulin-like growth factor 2(IGF2), known to play a crucial role in human growth and development. The IGF2 gene was selected for analysis because its methylation marks are stable up to adult age, making IGF2 gene a good candidate for such a study [11].
In this research, those subjects exposed to the Dutch famine during the first trimester had much lower IGF2 methylation levels compared to control unexposed individuals six decades after the hunger exposure. Subsequently, this observation has been extended by examination of a set of 15 additional candidate loci responsible for development of metabolic and cardiovascular phenotypes. Levels of methylation of six of these loci (GNASAS, IL10, LEP, ABCA1, INSIGF and MEG3) have been found to be associated with prenatal exposure to famine [6,11,13,14].
In 2013, a smaller subgroup of individuals (150 men and women) from the Dutch famine birth cohort (aged 68) were reported to display increased mortality and were more frequently admitted to hospital forage-related disease. In the same study, at the organ/system level, the authors demonstrated larger increases in levels of aging biomarkers in the brain, bone, skeletal muscle and the eye. At the cellular level, increased inflammation, oxidative stress and decreased telomere length of DNA, a hallmark of cellular senescence, were evident. Overall, this evidence has led to the emergence of the notion that, even decades later, organs, tissues and even cells can be programmed as a result of early-life fetal environmental encounters [12].
Findings from the Dutch Hunger Winter Study on the developmental origin of T2D were also confirmed in populations of other countries such as Austria. Based on the data set including 325,000 Austrian diabetic patients, investigators observed an excess risk of T2Din those persons who were born during or immediately after a famine episode. For instance, individuals born between 1919–1921 displayed up to 40% higher chances of having T2D in those compared to those who were born in 1918 or 1922 [11].
In this section, we examine several EWAS studies that leverage the resolution capacity of an omics systems approach to expanding our understanding of how nutritional environments impact health and disease (CVD).The Genetics of Lipid Lowering Drugs and Diet Network (GOLDN) study supplies data supporting several investigations. GOLDN was initiated to assess how genetic factors interact with environmental (diet and drug) interventions to influence blood levels of triglycerides and other atherogenic lipid species and inflammation markers. The study recruited a total of 1327 individuals and a total of 1053 met all eligibility requirements. Caucasian participants primarily from three-generational pedigrees from two NHLBI Family Heart Study (FHS) field centers (Minneapolis, MN and Salt Lake City, UT). Only families with at least two siblings were recruited and only participants who did not take lipid-lowering agents (pharmaceuticals or nutraceuticals) for at least 4 weeks prior to the initial visit were included. A total of 1048 GOLDN participants were included in the diet intervention [3].
The first is the GOLDN study that examines the epigenomic impact on inter-individual PPL variability. The study conducted a EWAS study on 979 subjects with DNA methylation measured from CD4+ T cells, who were challenged with a high-fat meal as a part of the GOLDN study. Postprandial lipemia (PPL), is the increased plasma triglyceride (TG) concentration after consuming a highfat meal, and is an independent risk factor for CVD. Individual responses to a meal high in fat vary greatly, depending on genetic and lifestyle factors. However, only a few loci have been associated with TG-PPL response. Heritable epigenomic changes may be significant contributors to the unexplained inter-individual PPL variability. Eight methylation sites encompassing five genes, LPP, CPT1A, APOA5, SREBF1, and ABCG1, were significantly associated with PPL response at an epigenome-wide level(P<1.1×10-7), but no methylation site reached epigenome wide significance after adjusting for baseline TG levels. Higher methylation at LPP, APOA5, SREBF1, and ABCG1, and lower methylation at CPT1A methylation were correlated with an increased TG-PPL response. Diet-induced epigenetic variation in obesity has been demonstrated recently and is transmissible in mice from one generation to next. These eight PPL-associatedmethylation sites, also correlated with fasting TG, account for a substantially greater amount of phenotypic variance (14.9%) in PPL and fasting TG (16.3%) when compared with the genetic contribution of loci identified by a previous GWAS (4.5%). In summary, the epigenomeis a large contributor to the variation in PPL, and this has the potential to be used to modulate PPL and reduce CVD [3,6,13-15].
The effects of macronutrient derivatives (e.g., methionine, choline, and betaine), and micronutrients such as vitamins (e.g., B-vitamins, D vitamin, and retinoic acid, microminerals or trace elements (e.g., iron, zinc and selenium) and bioactive compounds (e.g., phytochemicals including polyphenols) on epigenetic processes will be reviewed in brief. These dietary compounds are an integral part of everyday nutrition in human populations worldwide, are frequently present in maternal diets, and have been reported to influence epigenetic mechanisms. The phytochemicals lycopene in tomatoes, genistein in soybeans, resveratrol in grapes and berries, sulforaphane in broccoli, epigallocatechin-3-gallate (EGCG) in green tea, or curcumin and allyl sulfur compounds present in spices are among a growing list of agents used in the modern diet that might affect epigenetic mechanisms [8].
Over the past years, in vitro and in vivo studies have shown that polyphenols modulate the activity of cell signaling proteins, transcription factors and consequently the expression of both, mRNA and miRNA. In vitro, polyphenols impact the expression of genes in various cell types. It has been demonstrated that flavan one metabolites at physiological concentrations can modulate the expression of genes involved in inflammation in tumor necrosis factor (TNF)-and stimulated human umbilical vein endothelial cells (HUVECs). Exposure of human monocytes to green tea polyphenols affected the expression of genes related to atherosclerosis development, such as CD36, LXR-a, MYC or LDL-R. Inhuman hepatocytes polyphenols from red grape juice were able to counteract the LDL-induced changes in gene expression of the LDL receptor, hydroxymethylglutaryl-CoA (HMG-CoA) reductase and the transcription factor sterol regulatory element-binding protein (SREBP)-1, a key regulator of lipid homeostasis. Also genes such as ICAM1, MCP1, IL6 or IL1b of which expression is controlled by nuclear factor kappa B (NF-kB) and of which transcriptional activation is critical in a number of pathologies including cardiovascular diseases, are differentially expressed in the presence of polyphenols [8].
Currently, few studies describing the genomic impact of polyphenols and polyphenol-rich foods in humans have been published. Daily intake of 150 mg quercetin for 2 weeks significantly affected the expression of 788 genes in CD14-positivemonocytes, genes related to immune system, apoptosis or cell signaling pathways. Consumption of orange juice rich in hesperidin or hesperidin alone for 4 weeks by healthy, middle-aged, moderately overweight men significantly affected over 3,000and 1,800 genes respectively. These genes were involved in the regulation of chemot axis, cell adhesion, lipid transport and their expression profile could be considered as anti-inflammatory andanti-atherogenic. More recently, in a double-bind, randomized cross-over trial in menopausal women isoflavones affected in peripheral blood mononuclear cells the expression of 357 genes which are involved in inflammation, oxidative phosphorylation and cell cycle have been published up to now describing the genomic impact of polyphenols and polyphenol-rich foods in humans. It has been observed that the daily intake of 150 mg quercetin for 2 weeks significantly affected the expression of 788 genes in CD14-positivemonocytes, genes related to immune system, apoptosis or cell signaling pathways [16].
Several dietary epigenetic modulators have been investigated for further understanding of molecular mechanisms underpinning epigenetic effects in the context of chemoprevention. Some have shown potential to reverse methylation-induced silencing and change the expression of various genes (i.e., DNMT inhibitors). For example, EGCG of green tea extract is a demethylating agent that inhibits catechol-O-methyltransferase (COMT), the enzyme responsible for the inactivation of catechol molecules. This enzyme introduces the methyl group from AdoMet onto the catecholamine group forming AdoHcy, a potent inhibitor of DNMTs. On the other hand, EGCG can form hydrogen bonds with different residues in the catalytic pocket of DNMTs acting as a direct inhibitor of DNMT1.
Lycopene, a bright red carotene and carotenoid pigment found in tomatoes and other red fruits and vegetables modulates the expression of numerous genes relevant to cell cycle control; however, lycopene and apo-10’-lycopenal are not effective demethylating agents of GSTP1 in the human LNCaP prostate cancer cell line. It has been demonstrated that treatment of the MV4-11 leukemia cell line with curcumin decreases global DNA methylation. Genistein, a major phytoestrogen in soybeans, induces a dose-dependent inhibition of DNMTs. Furthermore, prenatal exposure to genistein affects fetal erythropoiesis and exerts lifelong alterations in gene expression and DNA methylation of hematopoietic cells. Sulforaphane, a bioactive component of cruciferous vegetables, down-regulates DNMT1 and induces demethylation of the Cyclin D2 (CCND2) gene in the human colorectal adenocarcinoma Caco-2 cell line. A wide variety of cruciferous vegetables contain isothiocyanate compounds, which are known to affect the epigenome. For example, isothiocyanates, metabolites of glucosinolates present in cruciferous vegetables, lead to demethylation and re-expression of GSTP1. Resveratrol, a polyphenol phytoalexin in grape peel and a weak inhibitor of DNMT activity, increases the ability of adenosine analogues to reduce DNA methylation and increase the expression of RARβ2 in human breast cancer MCF-7 cells [8].
A double-blind, randomized, placebo-controlled intervention study recently revealed, that daily consumption of monomericand oligomeric flavanols (MOF) derived from the seeds of grapes (VitisviniferaL.) for 8 weeks confer a vascular health benefit in male smokers. This potential cardiovascular health benefit is in agreement with a recent study that showed an association between the intake of flavanols and reduced risk of coronary heart diseases. However, comprehensive understanding of the biological activity of flavanols including their mechanisms of action in humans is still enigmatic [16]. The gene expression profile of blood leukocytes was obtained following 8 weeks consumption of MOF. Statistical analyses revealed 864differentially expressed genes (p.0.05 and corrected p,0.1). Out of the 864 genes, 445 were identified as down regulated and 419 as up-regulated, with expression changes varying from 1.32 to 1.72 [16].
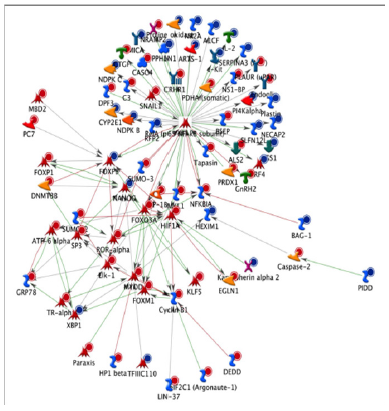
Figure 4: An NF-kB biological network based on the differentially expressed genes in circulatory cells obtained from MetaCoreTM network software and the ‘‘Analyze’’ network algorithms. Dots in the right corner of a gene indicate differential expression, blue for down regulated genes and red for up-regulated genes.
To identify the biological function in which these genes are involved, gene ontology, pathway and gene network analyses were performed. Gene ontology revealed that differentially expressed genes are involved in different over-represented cellular processes. Among these processes are those regulating macrophage-derived foam cell differentiation, chromatin organization, cell cycle, chemotaxis, regulation of cell-cell adhesion, regulation of metabolic processes, regulation of lipid storage and regulation of cytoskeleton organization. To further refine the biological functions in which differentially expressed genes are implicated, the genes were placed according to their role(s) in cellular ormetabolic pathways. Pathway analyses of the differentially expressed genes using MetaCore software identified 59 significantly over-represented cellular pathways. Pathway analyses of the differentially expressed genes using Meta Core software identified 59 significantly overrepresented cellular pathways. Among these 59 pathways, 19 are involved in the regulation of inflammation, cell adhesion, cell cycle and cytoskeleton remodeling. Pathway analysis using KEGG database also revealed pathways such as cell adhesion, MAPK signaling pathway, regulation of actin cytoskeleton, cell cycle, chemokinesignaling pathway, focal adhesion and leukocyte transcendothelial migration. Among the genes present in these pathways areCXCL12, SCRIB, PDGFRL, FERMT3, ICAM1 or VCAM1. Among the identified transcription factors are SP1, p53, AP1 and NF-kB. A network of genes centred to NF-kB and affected by the MOFsupplementation is presented in (Figure 4) [16].
Genetic factors contribute to the risk of complex metabolic diseases such as obesity and type 2 diabetes. Although genomewide association studies (GWAS) have identified numerous genetic loci influencing the risk of developing obesity and type 2 diabetes, only a few of these loci have been linked to the molecular mechanisms contributing to the phenotype outcome. However, the identified genetic loci only explain a modest proportion of the estimated heritability of these diseases. Additional genetic mechanisms remain to be characterized, and may include genetic variants interacting with epigenetic modifications. A study conducted by Volkov et al. [17] provides the first detailed map of genetic loci in both the cis and trans positions affecting the genomewide DNA methylation pattern in human adipose tissue and their association with numerous metabolic traits. The identified mQTLs cover known lipid, obesity and diabetes loci. It also highlights the interaction between genetic and epigenetic variation inadipose tissue and pancreatic islets to generate new insights into the biological processes affecting disease susceptibility [17]. mQTL Analysis in Human Adipose Tissue Identifies Genetic Variants Associated with Metabolic Traits
A summary of the research findings are presented here. Volkov et al. [17] performed a genome-wide DNA methylation quantitative trait locus (mQTL) analysis in human adipose tissue of 119 men, where 592,794 single nucleotide polymorphisms (SNPs) were related to DNA methylation of 477,891 CpG sites, covering 99% of RefSeq genes. SNPs in significant mQTLs were further related to gene expression in adipose tissue and obesity related traits. Volkov et al. [17] found 101,911 SNP-CpG pairs (mQTLs) in cis and 5,342 SNP-CpG pairs in trans showing significant associations between genotype and DNA methylation in adipose tissue after correction for multiple testing, where cis is defined as distance less than 500 kb between a SNP and CpG site.
This was the first time that a research team presented mQTL results in adipose tissue looking at both cis and trans effects in most genomic regions and genes [17].
These mQTLs include reported obesity, lipid and type 2 diabetes loci, e.g. ADCY3/POMC, APOA5, CETP, FADS2, GCKR, SORT1 and LEPR. Significant mQTLs were overrepresented in intergenic regions meanwhile underrepresented in promoter regions and CpG islands. Volkov et al. [17] further identified 635 SNPs in significant cis-mQTLs associated with expression of 86 genes in adipose tissue including CHRNA5, G6PC2, GPX7, RPL27A, THNSL2 and ZFP57. SNPs in significant mQTLs were also associated with body mass index (BMI), lipid traits and glucose and insulin levels in our study cohort and public available consortia data [17].
In order to provide further insights into mechanisms of genetic and epigenetic interaction and its impact on regulation of metabolic phenotypes, we used the CIT. Volkov et al. [17] discovered 39 significant mQTLs where DNA methylation represents the mediator between genetic loci and a metabolic trait. One of these mQTLs SNPs is associated with HDL regulation through DNA methylation of a CpG site annotated to MCM6. This is an MCM (minichromosome maintenance) complex gene that previously has been shown to affect total cholesterol levels. Among other genes identified in the CIT analysis were TCF19, which has been associated with type 1 diabetes through GWAS [72], and CAMK1D, which has been associated with type 2 diabetes. This supports the role of DNA methylation as a direct mediator between genetic variation and metabolic phenotypes. Importantly, the Causal Inference Test (CIT) demonstrates how genetic variants mediate their effects on metabolic traits (e.g. BMI, cholesterol, high-density lipoprotein (HDL), hemoglobin A1c (HbA1c) and homeostatic model assessment of insulin resistance (HOMA-IR)) via altered DNA methylation in human adipose tissue (Figure 5) [17].
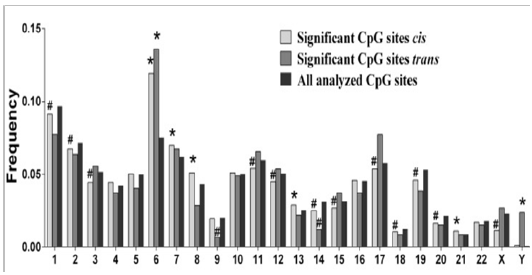
Figure 5: CpGcis and trans sites across the Genome. PLoS One. 2016; 11(6): e0157776.
Given that SNPs affect DNA methylation and that DNA methylation is a dynamic process that may change in response to environmental factors and affects phenotype transmission, it may be possible that the SNP effect on DNA methylation levels, and indirectly on metabolic phenotypes, may change under different environmental conditions [1-6,17]. It is hence possible that some of the identified mQTL SNPs overlapping with consortium data may have escaped detection to disease phenotypes in previous GWAS studies since DNA methylation levels was not considered. This form of gene-environment interactions could potentially affect the SNPs impact on disease risk. Indeed, our previous data, where we identified a SNP that introduces a CpG site in the promoter of NDUFB6, support this hypothesis.
Here, Volkov et al. [17] showed that while elderly carriers of the genotype that introduces a CpG site had a high degree of methylation in the SNP-CpG site together with decreased skeletal muscle NDUFB6 expression and decreased glucose uptake, young carriers had low degree of methylation in the SNP-CpG site together with increased skeletal muscle NDUFB6 expression and no effect on glucose uptake. Together, this study demonstrates a clear interaction between genetic, epigenetic and non-genetic factors. Additionally, genetic variation may carry inheritance of epigenetic variation and thereby have an impact on the heritability of human diseases and may explain some of the missing heritability of human complex diseases. Furthermore, Volkov et al. [17] also found that several SNPs associated with DNA methylation in adipose tissue overlapped directly or via proxy SNPs to previously reported disease loci of obesity related traits, including CETP and FADS2, which are both known to be associated with total cholesterol, LDL, HDL and triglyceride levels [18-20]. These data support the assertion that genetic and epigenetic variation work synergistically to influence metabolic phenotypes and disease risk in humans.
This review examines several innovative study designs that incorporate a combination of EWAS, GWAS, hap MAP, QTL sequencing and nutrigenomic investigative techniques to determine the risks to human health from changes in a dynamic nutritional environment. What we now understand, is that key developmental windows occurs across the human lifecycle. Additionally, the nutritional environment imprints epigenetic markers that not only temporally affect gene expression but also initiates adaptive responses involving multiple genetic networks underlying developmental programs that predisposes the individual to metabolic disease in the future.
Beginning with gamete biogenesis and maturation, embryonic development progresses through fetal development, and advances through the post-natal development phases that result in a mature individual. During early development is when the fetal epigenome displays a high degree of plasticity and is influenced by a variety of dietary compounds that contribute to the in-utero nutritional environment. For example, B vitamins support the development of the fetal cardiac and nervous system. Not surprisingly, current U.S. health policy even recommends that women with the capability of becoming pregnant consumea required amount of B vitamins daily. In this form, systems biology can be used to empower the myriad voices advocating for the prioritization of nutritional strategies to mitigate the impact of metabolic disease on human society. Nutrigenomics can be used as a policy tool to address the challenges of optimizing our global food chains to sustain population health outcomes. Firms introducing new food innovations into the market can leverage insights generated from EWAS and GWAS to design nutritional supplements based upon natural product chemistry to support personal nutrition.
The growing popularity of flavonoids, polyphenols and terpenes is one example of how scientific literature is influencing nutritional choices. Future studies are needed to expand our understanding of how combinations of micronutrients and macronutrients can be used to create nutritional environments that impact post-natal development. The possibility that we can design personalized diets with the intention of mitigating the epigenetic imprints introduced into the fetal epigenome is exciting. However, additional work examining human populations affected by famine, poverty, obesity and aging are needed.
The investigation by Volkov et al. [17] exploring the mechanism of SNPs interactions with DNA methylation sites revealed the highest deviation from expectation of CpGs insignificant cis-mQTLs was observed on chromosome 6 (p-value = 3.4x10-89), where the highlypolymorphic HLA region is located, a genomic region linked to numerous autoimmune diseases. This finding is significant because the HLA class II genes HLA-DRB1, -DQA1 and –DQB1 are the strongest genetic factors for type 1 diabetes. The implication of these findings is that the HLA may contain SNPs in significant mQTLs that may increase the risk of not only developing metabolic disease but also contributing to predisposition in future generations.

























