info@biomedres.us
+1 (502) 904-2126
One Westbrook Corporate Center, Suite 300, Westchester, IL 60154, USA
Site Map
Received: May 17, 2017 Published: May 31, 2017
Corresponding author: Hussam Abu Farsakh, First Medical Lab, Amman, P.O. Box: 2098, Al-Jubaiha, 11941, Jordan
DOI: 10.26717/BJSTR.2017.01.000115
We studied the morphological and histochemical changes in muscle biopsies from 6 cases with myasthenia gravis (MG), The patients were misdiagnosed at the initial presentation however they were all proved later by either histochmical and serological tests or improvement on mestinone therapy. Here, we described the changes seen in muscle biopsies of MG with atypical clinical presentation: atrophy of type II fibers; predominance of type I fibers; increased NADH staining at the neuromuscular junction (NMJ). The most consistent finding is marked reduction in acetylcholinesterease (AChE) activity at the end plates in all cases. The results of this study suggest that histochemical and acetylcholinesterase stains, in combination with clinical assessment can help to identify unsuspected cases of MG and can help to target the appropriate clinical investigations.
Keywords: Muscle biopsies; Myasthenia Gravis; Acetyl cholinesterase receptor
Myasthenia gravis is an autoimmune disorder of neuromuscular junction with the circulating antibodies against acetylcholine receptor (AchR) caused by auto antibodies against the nicotinic acetylcholine receptor on the postsynaptic membrane and characterized by variable weakness and fatigability of oculobulbar and limb muscles [1]. It has a bimodal peak of incidence with first peak in the third decade and the second peak in the sixth decade [2]. The diagnosis is usually made on clinical and electrophysiological grounds. In the majority of patients, initial presentation is due to the involvement of extra ocular muscles. With the progression of the disease, facial, bulbar, proximal limb muscles, neck extensors, and diaphragm get involved [3].
This disease has a prevalence of 2/10000 population [4]. Untreated MG has a 10 year mortality of 20-30% and 85% of the patients are seropositive for acetyl choline antibodies [5]. Currently very little is known about the histochemical changes of muscle fiber types in MG. This may be due to that most of the patients are diagnosed by clinical examination, serology, EMG and nerve conduction studies. However, in some cases, the clinical picture may not be typical and serology tests may either be negative or not available.
In this article, we are describing the muscle morphological and histochemical changes of MG. Knowing these changes are essential for neuropathogist so as not to miss cases with atypical presentation. We would like also to emphasize the value of routine use of acetylcholinesterease (AChE) staining in muscle biopsy.
We studied 6 patients (3.3% of 182) who visited The First medical Lab in Amman, Jordan, during a 5 year period. Muscle biopsies performed on 380 patients during the months of January 2000 through to September 2007. Age ranged from 11 to 60 years (5F, 1 M).
None of patients had a diagnosis of myasthenia gravis at the time they presented for muscle biopsies. Data were collected regarding muscle power, pattern of weakness, functional abilities, EMG studies, acetylcholine receptor antibodies.
Diagnostic needle muscle specimens were obtained from the quadriceps or the deltoid muscles of the six patients. Standard histological and histochemical analysis was performed for the specimens of all patients as described [6]. 10 μm serial crosssections were cut in a cryostat. Sections were stained with hematoxylin-eosin, modified trichrome stain, NADH tetrazolium reductase (NADH-TR), succinic dehydrogenase (SDH), myofibrillar ATPase at pH 10.6., 4.2, 4.6, and acetyl cholinesterase (AChE).
Table 1: Clinical details and laboratory features of the six patients.
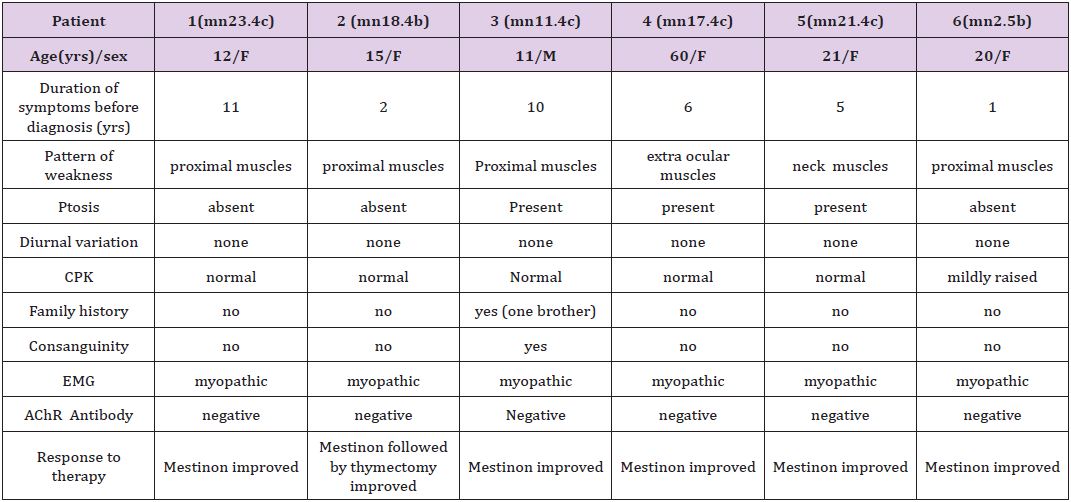
Table 1 show that we have two peaks of occurrences. One peak at the second decade: five patients (age 11-21 years), and another peak at the sixth decade one patient (age 60).
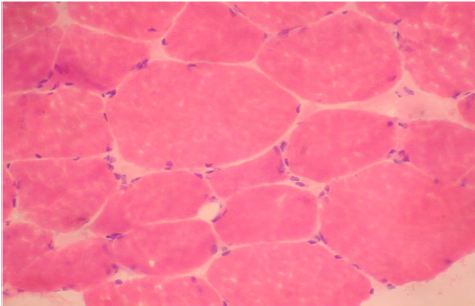
Figure 1: Quadriceps muscle biopsy of patient 1. Hematoxyline and eosin sections showing variation in fiber size with interspersed mildly atrophic fibers. The atrophic fibersis seen selectively in type II fibers. (Mag.400x).
Routine histological analysis of the quadriceps or deltoid muscle specimen of all cases showed mild variation of fiber size with interspersed mildly atrophic fibers (Figure 1). The abnormal variability of fiber size with type I fibers predominance was clearly demonstrated in Figure 2 and were seen in cases no. 1,4 &5. The histochemical examination of the muscle biopsies of all patients revealed marked reduction in ACHE activity (Figure 3). There were increased oxidative enzyme NADH reactions at the neuromuscular junction in all cases but case no. 3 (Figure 4). Table 2 shows the characteristics of the histochemical changes in six myasthenia gravis patients.

Figure 2: Quadriceps muscle biopsy of case 1 with alkaline preincubation ATPase stain at pH 10.6 showing predominance of type 1 fibers. (Mag.400).

Figure 3: Quadriceps muscle biopsy of case 2 showing marked reduction of A Chase activity (Mag. 400x).
Table 2: Summary of the morphological changes in MG patients.
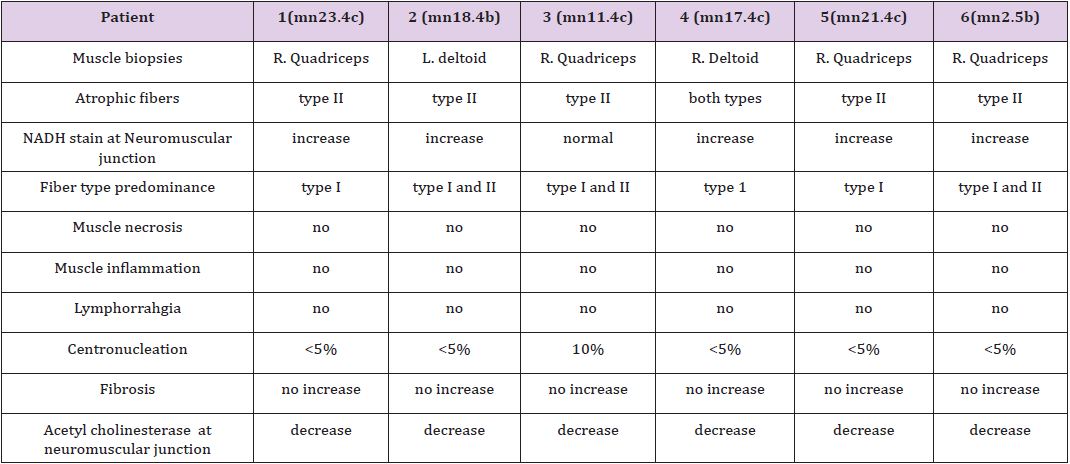
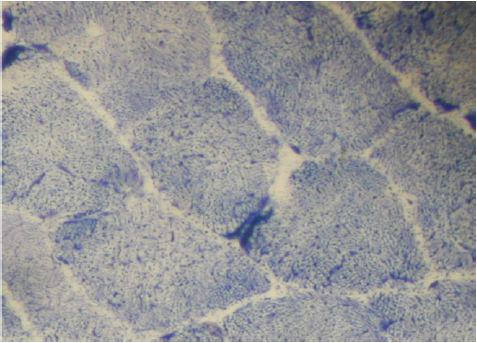
Figure 4: Quadriceps muscle biopsy of case 2 showing muscle fibers with increased NADH staining at the NMJ.
The histological picture in all cases showed almost common features of the following: minimal muscle fiber variation in size even in cases with long standing myasthenia gravis, and showed occasional atrophied fibers. In three of the six patients, predominance of type I fibers were observed, the atrophied fibers tended to be of type II. The most consistent finding in all cases was marked reduction in AChE staining at the end plates, However, increased NADH at the end plate were seen in five of the six patients. None of the cases showed lymphorrhagia that had been described before. In three of the six patients, electron microscopy was performed. In two of three cases mild increase in glycogen was noted and in one case, glycogen increase was coupled with increased fat globules.
AChR antibody was positive only in one case out of six cases (16%). All patient responded dramatically to AChI (like mestinon) and thymectomy was performed in case no.2. Thymic hyperplasia was seen on microscopic examination.
The six cases of myasthenia gravis reported in this study showed some features similar to those reported in the description by Dubowitz [6]. Except for Dubowitz description, the recent literature lacking significantly any other descriptive features of histochemical staining for MG. In our study, the six patients constitute 3.3% of 182 patients observed in a 5-year period. All patients represented a diagnostic challenge. For them, different diagnoses other than MG have been previously proposed, including myositis, motor neuron diseases, limb girdle muscular dystrophies, or mitochondrial myopathies. Understanding that MG can have atypical clinical presentation is very important for their diagnosis.. While 90% of patient with MG are reported to have AChR Antibody [7-9] patients with atypical clinical presentation that come to muscle biopsy may have much less chance of having positive AChR Antibody. In our series, only one out of six cases (16%) had positive AChR Antibody. In that case, the AChR Antibody was ordered by his clinician only after we suspected the diagnosis based on histochemical and AChE staining.
The inability of recognizing the morphological changes of MG by the pathologist, render diagnosing such cases correctly almost impossible. In our series, the duration of patients’ symptoms ranged from 1 year to 11 years. We strongly emphasize the use of AChE staining in any case that the pathologist sees minimal variation of muscle fibers disproportionate to the muscle power of the patient. The decreased AChE stunning has been reported before in Washington’s University web site (www.neuro.wustl.edu), and we agrees with their report.
Thymic hyperplasia is noted in 75% of patient s with MG. Of these germinal hyperplasia is noted in 85% and thymic tumor in 15%. Reference 1 Thymectomy, a known modality for the treatment of MG, induced excellent response in case no.2. None of the other cases had thymectomy. None of our patients had typical diurnal variation seen in MG with better power in the morning. Most of the affected muscles were the facial muscle, neck muscle and proximal upper and lower limb muscles. This selective pattern of muscle involvement may hypothesize a selective functional alteration of neuromuscular transmission [8]. The pathological mechanism leading to selective proximal muscle weakness and wasting in MG patients may be related to the presence of unknown auto antibodies or specific immunologic profile.
It is interesting to report that lymphorrhagia which has been described by Russell 1953, Adams et al. 1962, Engel and McFarlin, 1966 is not seen in any of our 6 cases and it is not a constant finding [6]. Dubowitz has emphasized the presence of type II atrophy with patchy distribution in 4 out of 6 cases of his study, he noticed type I fiber atrophy in some areas in 4 out of 6 cases. He suggested that the reason for these patchy changes were not all motor units myasthenia [6].
The increased NADH staining at the NMJ has been described by Rodolico et al as an increased tubular aggregates at the NMJ in patient with MG. (refrence 2 in my papers ) We observed this finding in 5 out of 6 six cases, and helped us is making the diagnosis in these cases. However, this findings is not specific and has been observed in cases other than MG in our entire cases.
In conclusion, MG is a common muscle disease, but it comes to muscle biopsy only rarely because most of these patients are diagnosed based on clinical and serology grounds only. Pathologists may miss the diagnosis of MG if he is not aware of the changes that occur in such cases or AChE staining is not available in his lab.

























