info@biomedres.us
+1 (502) 904-2126
One Westbrook Corporate Center, Suite 300, Westchester, IL 60154, USA
Site Map
Received: April 12, 2017 Published: May 25, 2017
Corresponding author: V Balaram, CSIR, National Geophysical Research Institute, Hyderabad – 500 007, India
DOI: 10.26717/BJSTR.2017.01.000109
EP: European Pharmacopoeia; USP: United States Pharmacopoeia; ICH: International Conference on Harmonization; EMA: European Medicines Agency; FDA: Food and Drug Administration; BP: British Pharmacopeia; JP: Japanese Pharmacopoeia; IP: Indian Pharmacopoeia; PDE: Permitted Daily Exposure
This brief review presents the international approaches to assessment of the content of geotaxis impurities (residual solvents and various inorganic and organic impurities) in pharmaceuticals. Nowadays, it has become necessary to provide not only purity profile but also impurity profile of a particular pharmaceutical product because of national and international regulations. These aspects along with significance of the quality, efficacy and safety of pharmaceuticals, including the source of impurities, kinds of impurities, control of impurities and regulatory aspects are discussed.
The supply of essential medicines of good quality has been identified as one of the prerequisites for the delivery of health care system of any country as poor quality medicines can harm or even kill consumers. The presence of unwanted chemicals in a particular medicine, even in extremely small quantities, may influence its efficacy and safety. Unlike in other industries, a medicine is a dynamic product whose color, consistency, weight, and even chemical identity can change between manufacture and ultimate consumption. Hence, quality of pharmaceuticals has been a concern of the people of the whole world, and is now receiving critical attention from regulatory authorities [1].
Impurities in pharmaceutical products are of great concern not only due to the inherent toxicity of certain contaminants, but also due to the adverse effect that contaminants may have on drug stability and shelf-life. In pharmaceutical and drug products, impurities are the unwanted chemicals (organic, inorganic and residual solvents) that remain with the active pharmaceutical ingredients (APIs), or develop/added during formulation, or upon aging. Organic impurities are the most common impurities found in every API which get incorporated normally during the multi-step synthesis process despite proper care [2,3]. Some of the metals such as lead, arsenic and platinum even at low concentrations are toxic and harmful to humans [4]. The presence of these organic and metallic impurities even in small amounts may influence the efficacy and safety of the pharmaceutical products. Therefore, impurity levels need to be controlled such that pharmaceutical products are sufficiently safe to be administered to humans.
The recent changes in the European Pharmacopoeia (EP), the United States Pharmacopoeia (USP) and the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) regulations for inorganic impurities and new strategies require companies to adopt new strategies for heavy metal analyses. The European medicines agency (EMA) has set guidelines for the limits of residual metal elements. The U.S. Food and Drug Administration (FDA) and the British Pharmacopeia (BP) and other regulating bodies such as the Japanese Pharmacopoeia (JP) and the Indian Pharmacopoeia (IP), strongly advise that contamination problems should be fully investigated in a timely fashion. The existing ICH Q3A Guideline classifies impurities as organic, inorganic, and residual solvents [5]. The Q3A and Q3B Guidelines effectively address the requirements for organic impurities, while Q3C covers requirements for residual solvents. The proposed new Guideline, Q3D, would provide clarification on24 elemental impurity (Cd, Pb, As, Hg, Co, V, Ni, Tl, Au, Pd, Ir, Os, Rh, Ru, Se, Ag, Pt, Li, Sb, Ba, Mo, Cu, Sn, and Cr) requirements are specified with their toxicity limits, defined as maximum permitted daily exposure (PDE) levels in μg/ day for the four major drug delivery categories. This paper presents a short review of the application of various modern analytical techniques for the accurate determination of impurities in APIs and pharmaceutical products with more emphasis on inorganic impurities.
Quality control (QC) and quality assurance (QA) in a pharmaceutical industry is the process of verifying and ensuring the quality of the product and to check whether the products meet specifications and will satisfy requirement. These procedures also ensure that the impurities (both organic and inorganic) are below acceptable limits and the label claim is correct. The purpose of these quality protocols in pharmaceutical industry is to help and ensure that each medicine reaching a patient is safe, effective and of acceptable quality.
Inorganic impurities such as As, Cd, Cu, Sn, Sb, Pb, Bi, Ag, Hg, Mo, In, Os, Pd, Pt, Rh, Ru, Cr, Ni and V in pharmaceutical-drug substances / products may originate from various sources like metal catalysts and metal reagents used during the synthesis of an active pharmaceutical substance (e.g., from naturally derived plant or mineral sources) and the excipients (e.g., stabilizers, fillers, binders, release agents, flavors, colors and coatings), impurities from manufacturing equipment (e.g., leaching from pipes), water and the container closure system (Figure 1). Analysis of impurities can be very challenging because they must be detected and determined at μg/g and ng/g levels. The quality of partially completed products are analyzed to determine if production processes are functioning properly. The heterogeneous physicochemical properties of pharmaceutical and drug samples, diversity of the matrices and divergent statutory requirements impose very high demands on the analytical laboratory procedures. Recent advances in analytical instruments have led to the development of techniques that allow us to increase the speed and amount of information collected on pharmaceutical samples.
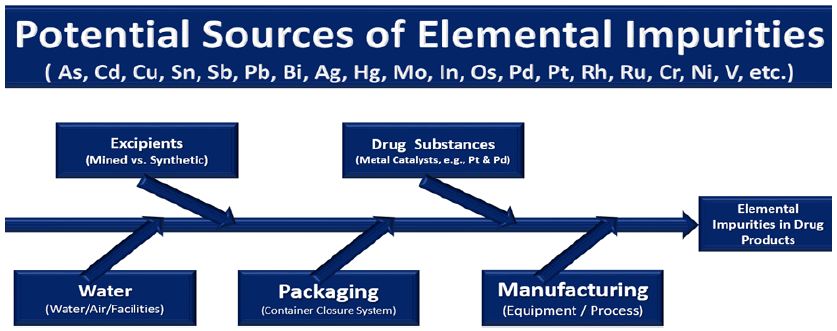
Figure 1: Potential sources of metallic impurities during the production process of drugs and pharmaceuticals.
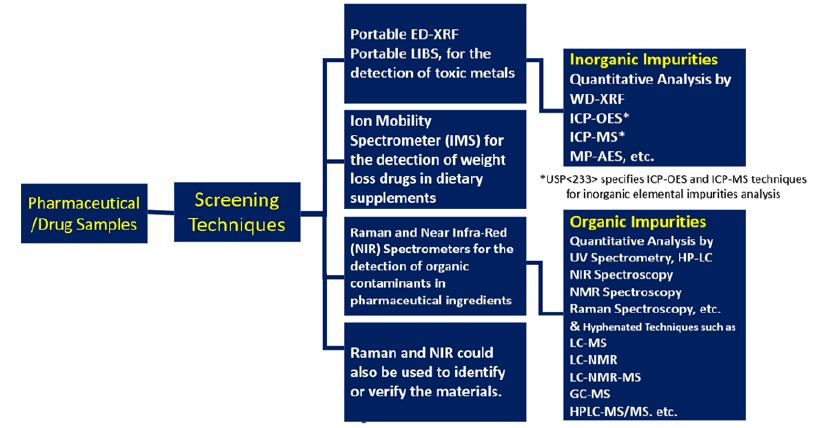
Figure 2: Summary of various analytical techniques used for the semi-quantitative and quantitative measurement of both organic and inorganic impurities during the production process of drugs and pharmaceuticals.
Such low levels require not only more sensitive analytical instruments, but also place higher demands on selectivity, because a higher number of other impurities may be present at lower concentration ranges. Until recently, inorganic impurities were being tested by a 110-year old qualitative method (USP < 231) which is not adequate for the purpose of controlling levels of potentially toxic elements, and consequently needs to be replaced by high sensitive instrumental methods such as inductively coupled plasma atomic emission spectrometry (ICP-AES) and inductively coupled plasma mass spectrometry (ICP-MS) which have the capability to carry out rapid and accurate multi-element analysis at and below ng/g levels coupled with excellent performance characteristics such as wide elemental coverage, rapid analysis (all elements at once), wide analytical working range (up to 9 orders), simple spectra and high matrix tolerance. In fact, both ICP-AES and ICP-MS have been recently recommended in the USP <233> chapter. Alternative technologies, such as wavelength dispersive x-ray fluorescence spectrometry (WD-XRF), atomic absorption spectrometry (AAS - both flame and graphite furnace) and microwave plasma atomic emission spectrometry (MP-AES) may be used, provided validation requirements are met. Very high sensitive analytical technique high resolution inductively coupled plasma mass spectrometry (HR-ICPMS) might find its utility in future when more number of elements such as all platinum group elements and rare earth elements [6] are added at lower threshold limits to the ICH list [1]. Figure 2 presents a summary of various analytical techniques used for the semi-quantitative and quantitative measurement of both organic and inorganic impurities during the production process of drugs and pharmaceuticals.
Sample decomposition methods play a very important role in these investigations. Possible methods are closed and openvessel digestion procedures. In case of digestion of volatile elements such as zinc, cadmium and mercury, open-vessel methods are not recommendable. To identify the right method, spike recovery experiment should be performed. Clear solution is acceptance criteria. Savillex fluoropolymer (PFA and FEP) pressure decomposition vessels are ideally suited due to their chemical inertness, wide working temperature range (-200oC to 260oC) and cleanliness.
In addition, quality control protocols involving measurement against appropriate and traceable international certified reference materials (CRMs) are also required for testing the reliability of each batch of data produced in the laboratory. Since pharmaceutical CRMs for inorganic impurities are not easily available, many workers prepare synthetic calibration standards for quantitation purposes. When the calibration standard is introduced to the instrument, it must ideally be in a physical and chemical form that is sufficiently similar to the final form of the prepared test sample so that any errors in the analytical result due to matrix effects are negligible. Varieties of reference materials are available from international organizations such as National Institute of Standards and Technology (NIST), US, and a few other international organizations which are traceable to NIST standards.
For the medicine to serve its intended purpose they should be free from impurity or other interference which might harm humans. A medicine that passes all laboratory tests upon receipt may be useless within a few months if the packaging, storage, and transportation conditions are not maintained properly. The analytical techniques described here play a vital role in impurity profiling of pharmaceuticals from identification to the final structure elucidation of unknown impurities. It is necessary to use sensitive and specific analytical methods for detecting genotoxic impurities in drugs and pharmaceuticals. Reliable and accurate data measured in pharmaceutical laboratories are important to ensure that only safe and efficient drugs are authorized for marketing, and released for product shipment. Therefore, pharmaceutical development and QC laboratories have to follow international regulations to demonstrate data quality.

























