 Research Article
Research ArticleABSTRACT
Congenital myasthenic syndrome (CMS) is a neuromuscular disorder caused by pathogenic variants in several genes encoding junction proteins. One of these genes is CHAT which encodes choline acetyltransfer-ase. Here, we report a family with compound heterozygote CHAT disease-causing mutations. The c.1061C>T (p.Thr354Met) mutation has been known previously as a pathogenic DNA variant that is associated with CMS, while the c.619C>G (p.Arg207Gly) was deposed as a clinical variant with unproven pathogenicity. Both parents as well as their child have one of the heterozygous CHAT mutations and no clinical features. Meanwhile, two children harbored the compound heterozygous CHAT mutations and, consequently, severe clinical features incompatible with their lives. This case provides evidence for the pathogenetic nature of c.619C>G and extends the genetic spectrum of the CHAT gene mutations.
Keywords: CHAT; Congenital Myasthenic Syndromes; Genetic Diagnosis; Compound Heterozygous Mutation
Introduction
Congenital myasthenic syndrome (CMS) is a neuromuscular disorder caused by pathogenic variants in several genes encoding junction proteins. CMS has been recognized as a clinical syndrome for more than 40 years by now. About 5% of the total cases of CMS are related to CHAT gene, where there are about 50 different patho-genic mutations up to date, including deletions, duplications, missense, nonsense, and frameshift variations. Almost all of them are inherited as compound heterozygotes. Some mutations were found in both homozygous and compound heterozygous modes, other ones never did. With wide spreading of the NGS methods a lot of new variants of nucleotide substitutions can be found in CHAT gene. Some of them have been discovered for the first time as new ones, other ones were deposed as a clinical variant with unproven pathogenicity. Some of them have been known previously as a pathogenic DNA variant that is associated with CMS. Here, we presented the case of two newborn girls from Russia with compound heterozygous CHAT variants, including missense paternal c.619C>G (p.Arg207Gly) and missense maternal c.1061C>T (p.Thr354Met). Both pathogenic variants manifested as severe CMS which was incompatible with their lives.
Materials and Methods
Medical History
The index patient was the 3rd child of Russian parents. The 1st child had been suffering from the same symp-toms and died on her first day of life form hypoplasia of the lungs. The 2nd child was healthy at three years of age (by the day of submission, she is thirteen and she has no clinical features linked with CHAT mutations). The index patient was a girl born term with 3150g, APGAR 7/2. She showed respiratory insufficiency at birth and required immediate mechanical ventilation. Since then, she had continuously been having mechanical ventilation due to respiratory insufficiency, extubating attempts had been without success. In the neonatal period, she presented hypotonia of the muscles with myoclonic twitches of the limbs and the chest, as well as generalized myoclonic convulsions diagnosed as epilepsy. In the further course, she showed contractures in the knee joint and clones of the bulbar muscles. EEG was abnormal and a brain CT showed atrophy of the cerebral cortex with hydrocephalus. At the age of 4 months, a tracheostomy was performed to facilitate mechanical ventilation. She had recurrent pneumonia. At the age of 5 months, the girl was shifted from St. Petersburg (Russia) to the pediatric intensive care unit of the Center for Child and Adolescent Medicine at the University Hospital Heidelberg (Germany) for diagnostic evaluation. At that time, she presented with the clinical picture of a severe neuromuscular disorder dominated by generalized hypotonia, ocular, motor and bulb facial weakness and respiratory insufficiency with mechanical ventilation.
There was an absence of cough, light or corneal reflexes. Her motions were weak, movements were undirected and spontaneous motor function occurred just in the palms and digits. Laboratory examination at admission was unremarkable except for a mild normocytic, normochromic anaemia. Intensive neuro-metabolic diagnostics were performed with unremarkable or nonspecific findings, including lactate and ammonia in the blood, acylcarnitines in dried blood spots, amino acids in plasma, very long-chain fatty acids in plasma, lysosomal enzymes in plasma and leukocytes and organic acids in urine. Cardiac MRI showed a marked deficit of white matter with a thin corpus callosum and subdural hygromas on both sides, but no specific finding suggestive of a neurometabolic disease. Compared to previous images, white mat-ter loss was progressive. As the neurometabolic investigations were without any specific findings, whole-exome sequencing was initiated. At the age of 6 months, the patient was shifted back to hometown, where supportive treatment continued. She died before 1 years old. Whole exome sequencing revealed two heterozygous pathogenic variants in the gene CHAT, and the diagnosis of the congenital myasthenic syndrome was put at the age of 12 months.
Whole Exome Sequencing
Whole exome sequencing (WES) was performed on genomic DNA of the affected individual III16 as described previously. Coding DNA regions were enriched using the Sure Select Human All Exon 50Mb V5 and sequenced on a HiSeq4000 System (Illumina, CA, USA). Sequencing reads were aligned to the human GRCh37. Results of WES revealed III16 to be compound heterozygous for two distinct missense variants on the paternal and ma-ternal allele in CHAT [RefSeq accession number NM_020549.4]: c.[619C>G]; [1061C>T], p.[Arg207Gly]; [Thr354Met]. Both identified variants were confirmed by Sanger sequencing. Segregation analysis via Sanger sequencing confirmed the compound heterozygote status in the affected sibling (III14). Whereas the unaffected sister (III15) and parents were heterozygous for only one variant. Both identified variants are not pre-sent in the exome database of the Institute of Human Genetics, TUM Munich, containing more than 24,000 samples. Both variants are reported in gnomAD with a minor allele frequency of c.[619C>G] MAF = 0.000007954 and c.[1061C>T] MAF = 0.00002476. Both missense variants are predicted to be damaged by pph2, Sift and CADD bioinformatic prediction tools.
PGT-M Design
Planning the next pregnancy, the couple decided to perform IVF with PGT-M. Muscle samples from the pro-band, paraffin blocks from the affected sibs and blood samples from the parents, healthy child and eleven other relatives were collected. The genomic DNA was isolated using standard commercial kits. To detect the familial pathogenic nucleotide variants c.619C>G and c.1601C>T four primers were designed based on the sequence of the CHAT gene:
exon 4 for 5’-gct gtc agg atg gga ctg tt-3’ (49620437-49620456; NC_000010.11)
exon 4 rev 5’-atg agc tag gag cac caa gg-3’(49620639-49620620; NC_000010.11)
exon 7 for 5’- ttg acc tgt tta ccc cac agt-3’ (49627588-49627608; NC_000010.11)
exon 7 rev 5’- cac gag cat gag atg ggc at-3’ (49627832-49627813; NC_000010.11)
The PCR of exons 4 and 7 of the CHAT gene was performed under standard conditions. Sanger sequencing of exons 4 and 7 was carried out using an ABI 3500 DNA analyzer (Applied Biosystems). For the purpose of indirect DNA diagnosis, nine Short Tandem Repeat (STR) markers closely linked to the CHAT gene were chosen according to the GeneLoc database (Supplementary Table 1). Multiplex PCR of all STR-loci was carried out. The forward primers were labelled with fluorescent dyes and PCR products were analyzed on an ABI 3130 genetic analyzer.
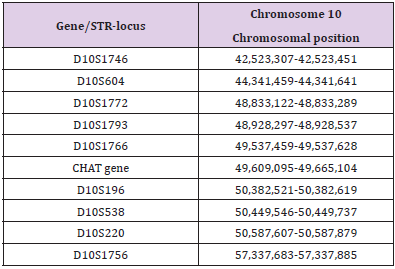
Supplementary Table 1: STR-markers linked to the CHAT gene.
Ovarian Stimulation Protocol
For ovarian stimulation recombinant follitropin alfa 225 IU pro day for 10 days, subcutaneous injections were used. For prevention of spontaneous LH-surge Ganirelix 0,25 mg per day from the size of the leading follicle more than 15 mm - for 3 days subcutaneous injections were used. For triggering of final oocyte maturation human chorionic gonadotropin 10000 IU subcutaneous injection was used. Transvaginal oocyte pick-up was after 36,5 hours from triggering. During pick-up 17 oocytes were received.
Embryo Culture Protocol
Every step of biomaterial relocation in the embryology laboratory was confirmed by an electronic witnessing system. Received oocyte-cumulus complexes were cultured at 37 ̊C, 6% CO2 and 3% O2 in bicarbonate-buffered cul-ture medium (G-IVF Plus, Vitrolife) under cultural mineral oil (Ovoil, Vitrolife) before denudation and fertiliza-tion. There were 16 mature (MII) and 1 immature (GV) oocytes. ICSI was performed in MOPS-buffered medi-um (G-MOPS Plus, Vitrolife). For the culture system, the Embryoscope incubator (Vitrolife) was used. Following ICSI, oocytes were individu-ally loaded into the micro-wells of the Embryoscope culture dish (Embryo slide, Vitrolife) filled with 25 ml pre-equilibrated 1-step culture medium (G-TL, Vitrolife) and covered by cultural mineral oil (Ovoil, Vitrolife). There were 8 normally fertilized oocytes in total and 8 oocytes degraded. The further incubation was uninterrupted until Day 6. Pictures of each embryo were taken by the time-lapse incubator in seven focal levels every 10 minutes. Eight blastocysts were developed feasible for the trophectoderm biopsy by Day 5 and 6 in total. The biopsy dishes with micro-drops of pre-equilibrated medium (CSCM-C, Irvine) under oil (Ovoil, Vitrolife) were prepared by a number of the embryos. Trophectoderm biopsy was performed according to the standard protocol. The samples include 6-7 trophectoderm cells. The blastocysts were vitrified (VT601, Kitazato) while the samples were sent for genetic testing.
PGT-M
The five-day embryos were biopsied and thophectoderma’s biopsies were collected. The whole genome ampli-fication (WGA) was performed using IonReproSeq PGS Kits (Thermofisher Scientific) according to the manu-facturer’s protocol. The DNA obtained from embryos were analyzed for c.619C>G and c.1601C>T changes and nine selected STR-markers. Five embryos were diagnosed as carriers of the maternal pathogenic allele c.1601C>T. In one embryo both pathogenic alleles were detected. Three embryos were diagnosed as healthy. No crossing over phenomenon was observed with any STR-marker located closer than 4Mb to the 5’-and 3’- ends of the CHAT gene.
PGT-A
For three healthy embryos and two carriers, PGT-A was carried out. The library preparation was performed with the use of the ReproSeq™ PGS Kit with Ion 520™ Chips (Thermo Fisher Scientific) according to the manufac-turer’s protocol. The sequencing was conducted on Ion S5 Semiconductor Sequencer (Thermo Fisher Scientific). Data analysis was performed with Ion Reporter™ Server System. In embryo #3 monosomy of chromosome 15 was detected and embryos ##1, 2, 5, 7 were euploid. Two independent transfers of male carrier embryos were performed in a three-month period. Unfortunately, pregnancy did not occur in any case.
Results and Discussion
According to our knowledge, there are 49 different pathogenic mutations in CHAT gene (Supplementary Table 2) including deletions, duplications, missense, nonsense, and frameshift variations. The 41 out of them are inherited as compound heterozygotes. Some mutations were found in both homozygous and compound heterozygous modes, other ones never did. For example, c.406G>A was found once in homozygous mode Arredondo, et al. [1], and thrice as compound heterozygous Shen, et al. [2]. The same is for c.1669G>A, which was found once as homozygous and thrice in compound heterozygous mode Shen, et al. [2]. The same case describes the family harbored c.406G>A and c.1669G>A simultaneously. The c.1249G>A was also found in both homozygous and compound heterozygous modes. Here we describe the new compound heterozygous variant consisting of c.1061C>T and c.619C>G. The c.1061C>T mutation is known previously as a pathogenic DNA variant that is associated with CMS, while the c.619C>G was deposed as a clinical variant with unproven pathogenicity. Both parents as well as their child have one of the heterozygous CHAT mutations and no clinical features (Figure 1). The c.1061C>T is located in exon 7 and changes an amino acid in the protein p.Thr354Met. According to Polychen2 HumDiv score, Poly-chen2 HumVar score and SIFT score the mutation is estimated as a pathogenic DNA variant (Supplementary Tables 3 & 4).
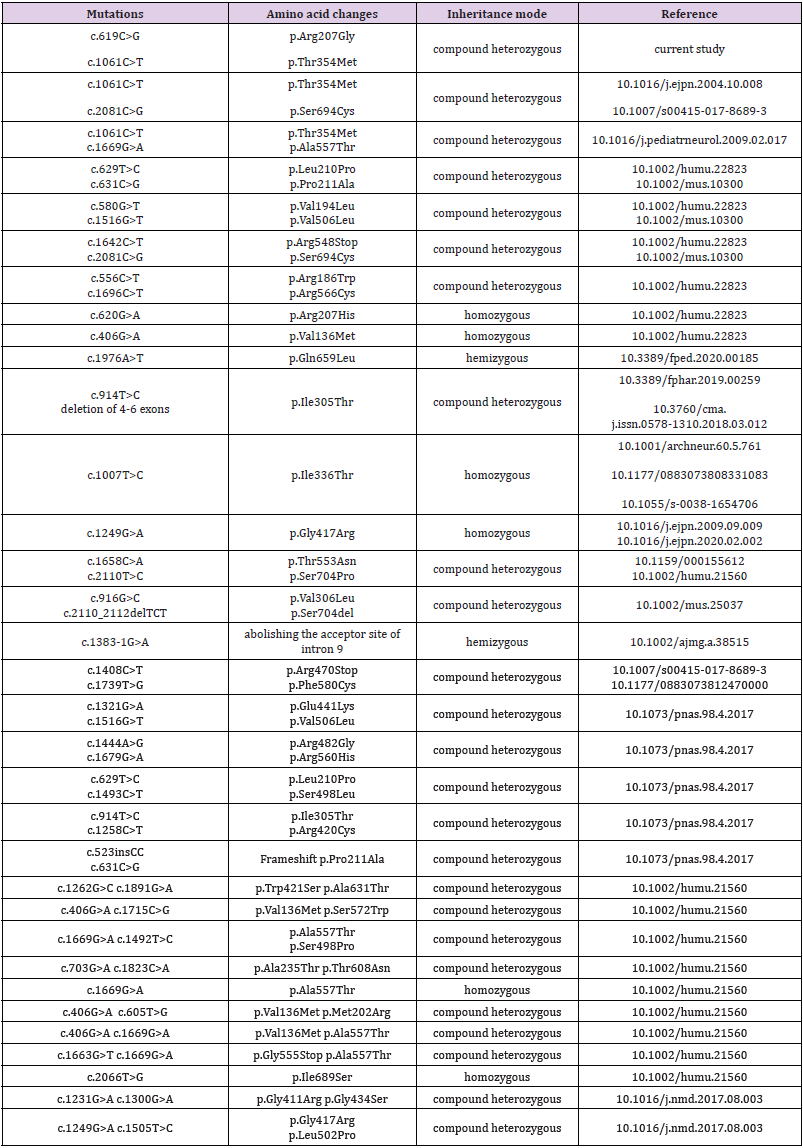
Supplementary Table 2: All known CHAT gene mutations.
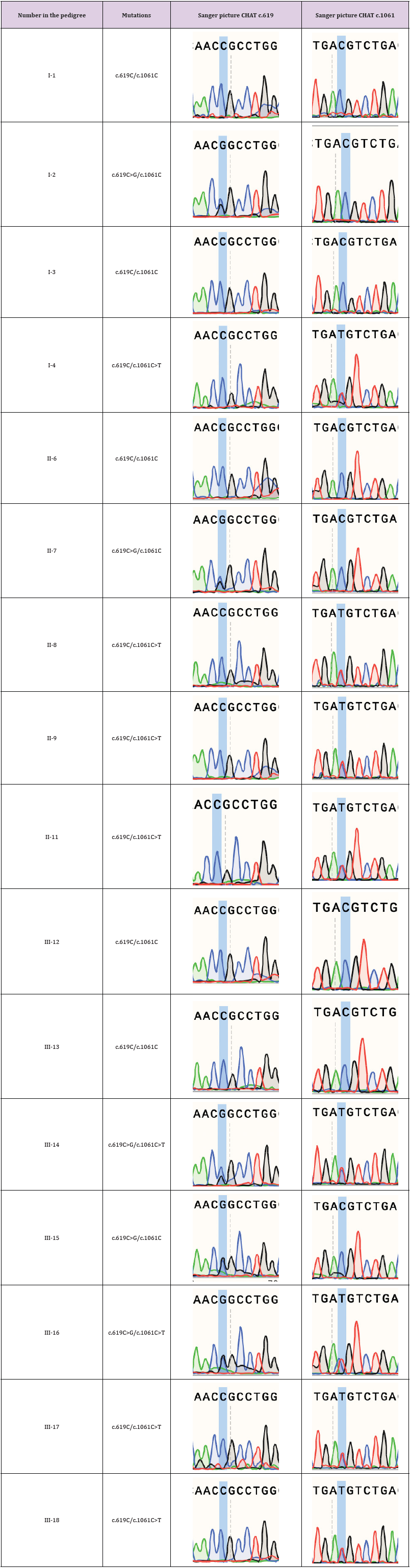
Supplementary Table 3: Mutations in the pedigree.
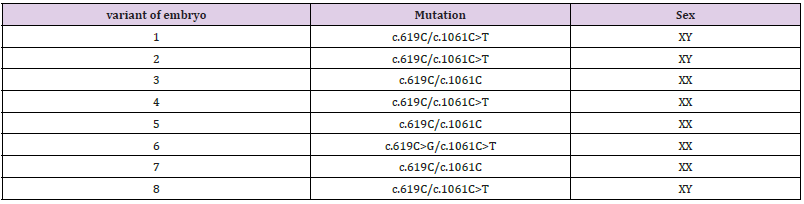
Supplementary Table 4: Mutations in embryos.
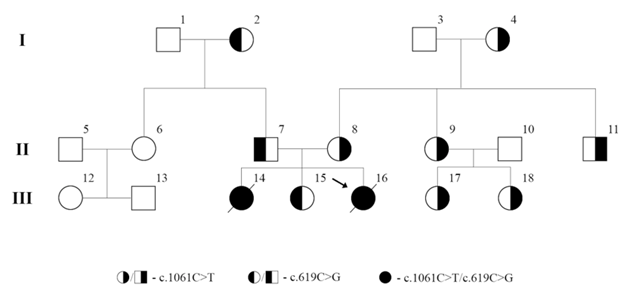
Figure 1: Pedigree of family under study. The two affected individuals III14 and III16 are compound hetero-zygotes for the paternal c.619C>G and the maternal c.1061C>T CHAT mutations. Both mutations extend back at least two generations. All mutations are presented in (Supplementary Table 2).
The c.619C>G that is in exon 4 and changes amino acid from Arg to Gly was deposed as a clinical variant with unproven pathogenicity. There are c.620G>A in the same codon in exon 4 that changes amino acid from Arg to His. The mutation was found in homozygous mode in affected patients Arredondo, et al. [1]. The variant с.619C>G of CHAT gene had not been reported in the ExAC (http://exac.broadinstitute. org/) or gnomAD databases (http://gnomad.broadinstitute. org/about). The variant c.1061C>T has an allele frequency of 2.48e-5 reported in ExAC and of 8.24e-6 in gnomAD. Both variants were predicted to be pathogenic by mul-tiple prediction software: PMut (http://mmb.pcb.ub.es/PMut/), SIFT (https://sift.bii.a-star. edu.sg/www/SIFT_seq_submit2.html), PolyPhen2 (http://genetics. bwh.harvard.edu/pph2/), Mutatu-onTaster (http://www.mutationtaster. org/), Fathmm (http://fathmm.biocompute.org.uk/) and CADD (https://cadd.gs.washington.edu/) (Table 1). The alignment of amino acid sequences revealed that arginine at position 207 as well as threonine at position 354 are highly conserved in different species (https://www.ebi.ac.uk/Tools/msa/), which suggests p.Arg207Gly and p.Thr354Met are likely pathogenic (Figures 2A & 2B). Three-dimensional structures of the wild-type and mutant CHAT protein of humans were con-structed by the homologous modelling database SWISS-MODEL (http://swissmodel.expasy.org/).
Residual changes between the wild-type and mutant CHAT proteins were mapped to the model by PyMol v2.4 software. This mutation causes a structural change in the substrate-binding site (Figures 2C-2E). Both mutations are located near the active site which assumes that they can disrupt the structure and catalytic activity of CHAT protein. Currently, there are several families where these mutations in compound heterozygous mode have been documented. Barictic, et al. [3] published two families with identical compound heterozy-gous mutations, c.2081C>G and c.1061C>T. Both probands having the mutations manifested intermittent apneas since early infancy. Another family published by Mallory, et al. [4] had com-pound heterozygous mutations, c.1061C>T and c.1669G>A. Proband had also severe episodes of apnea. In the case report was published a pedigree with four generations. The c.1061C>T was inherited from the maternal line and passed down from great-grandmother through a grandmother and then the father of the proband. The grandmother had eight children, six of them harbored the mutation in heterozygous mode. The c.1669G>A was inherited from the paternal line and passed down from grandfather through mother to proband. Here we also present the pedigree consisting of sixteen people. We sequenced exons 4 and 7 in CHAT gene for each person from the pedigree and showed the way the mutations are spent through the pedigree. There is no known consanguinity in the family.

Figure 2: Arginine (R) at a position 207 (A) and Threonine (T) at a position 354 (B) are highly conserved in CHAT in various species (the box marks the amino acid of interest). (C, D, E) Homology models for CHAT pro-tein generated using the crystal structures of CHAT protein (PDB accession code: 2FY4) as the template. The green cartoon represents the wild-type protein, the magnetic cartoon (C) represents the mutant protein c.619C>G (p.Arg207Gly), the pink cartoon (D) represents the mutant protein c.1061C>T (p.Thr354Met) and the orange cartoon (E) represents the protein with both mutations. Yellow dots represent polar contacts.
As can be seen from (Figure 1), the c.1061C>T passed down from grandmother through mother to probands. Grandmother passed down the mutation to all her children (two daughters and a son). One of the daughters passed down the mutation to both her daughters, while another one passed down to two out of three daughters. Eight people out of sixteen in pedigree harbors the mutation c.1061C>T. One but all were female. The c.619C>G also passed down grandmother through father to probands. One of the daughters who has no clinical features harbors the same mutation as her father. For indirect DNA diagnostics, STR-loci located in close proximity to CHAT gene were selected (Supplementary Figure 1). No family member showed a case of recombination between CHAT gene and markers located closer than 4Mb both distal and proximal to CHAT gene. This allows us to consider the selected STR-loci as suitable for additional information about the inheritance of the normal or mutant parental chromosome. Nor was it a surprise that cases of recombination were observed for markers located at a distance of more than 4Mb from the gene. Moreover, most of the crossing over was observed on the maternal chromosome: six times proximally to CHAT gene and once distally. Wherein, recombination on the paternal chromosome was observed only two times in the proximal side of CHAT. Just one more family member also had a case of recombination from the distal side of CHAT.
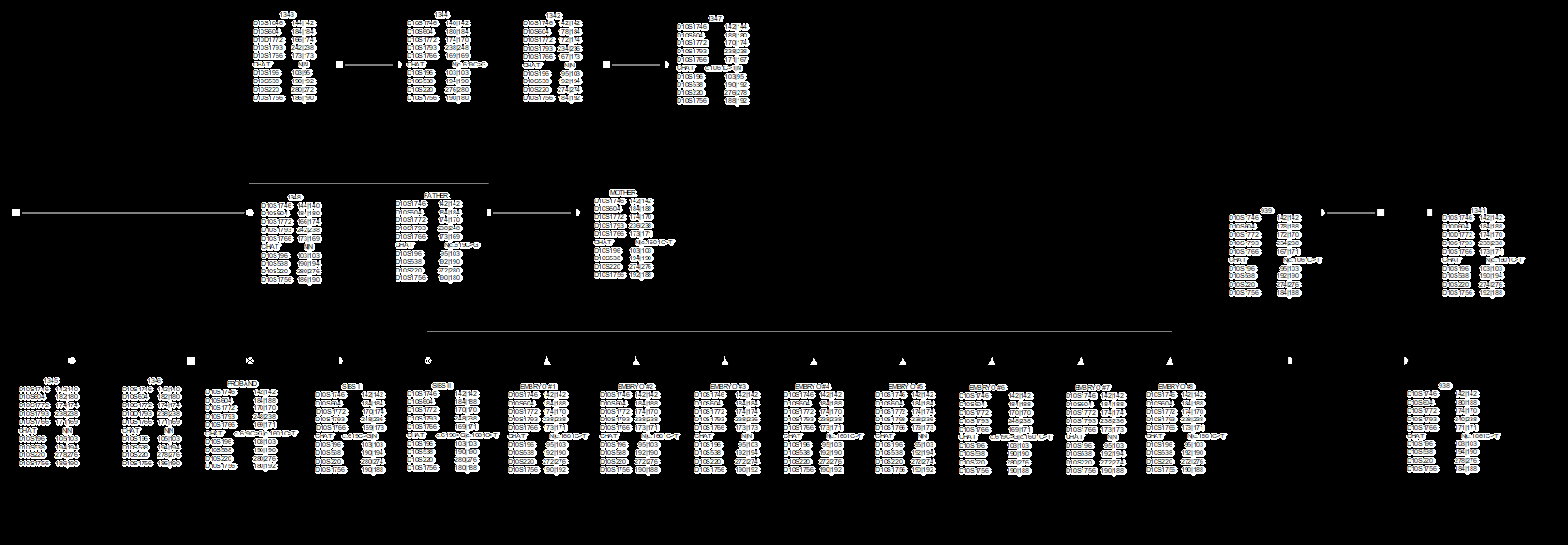
Supplementary Figure 1.
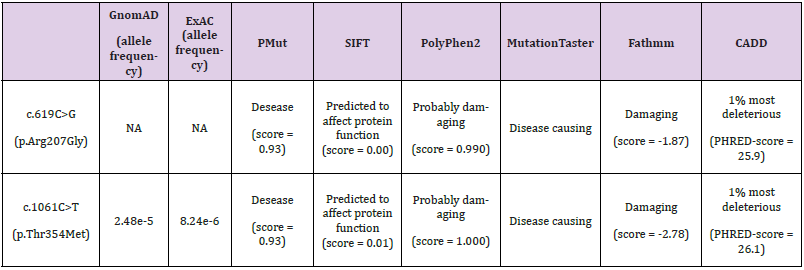
Table 1: In silico analysis of CHAT mutations described in this study.
In this work, we only state the fact that there are cases of crossing over, although it is possible that the high frequency of crossing over in the mother may not be accidental. However, this certainly requires sepa-rate studies. The couple reported here is currently underway the IVF protocol. After ovarian stimulation protocol, seventeen oocytes were received and fourteen were normally fertilized oocytes. Eight oocytes have grown to the blastocyst stage and were checked for both CHAT gene mutations. Half of the embryos were c.1061C>T in heterozy-gous mode, one of them was compound heterozygous mutations, and three were clear of the mutations. As we can see, the c.1061C>T is still in prevalence. According to our knowledge, there are no families with c.1061C>T alone and c.619C>G as well. We conclude that c.1061C>T spreads more aggressively through pedigree than c.619C>G due to unknown reasons and manifests itself only in compound heterozygous mode. We also specu-late that c.619C>G could be the pathogenic variant as well.
Supplementary Materials
Statements and Declarations
The authors declare no conflict of interest.
References
- Arredondo J, Lara M, Gospe SMJr, Mazia CG, Vaccarezza M, et al. (2015) Choline Acetyltransferase Mutations Causing Congenital Myasthenic Syndrome: Molecular Findings and Genotype-Phenotype Correlations. Hum Mutat 36: 881-893.
- Shen XM, Crawford TO, Brengman J, Acsadi G, Iannaconne S, et al. (2011) Functional consequences and structural interpretation of mutations of human choline acetyltransferase. Hum. Mutat 32: 1259-1267.
- Barisic N, Müller JS, Paucic-Kirincic E, Gazdik M, Lah-Tomulic K, et al. (2005) Clinical variability of CMS-EA (congenital myasthenic syndrome with episodic apnea) due to identical CHAT mutations in two infants. Eur J Paediatr Neurol 9: 7-12.
- Mallory LA, Shaw JG, Burgess SL, Estrella E, Nurko S, et al. (2009) Congenital myasthenic syndrome with episodic apnea. Pediatr Neurol 41: 42-45.
