 Research Article
Research ArticleABSTRACT
Wild-type (L10) and mutant strains AI3 and 3B of Pseudomonas aeruginosa have been widely used for production of native and altered aliphatic amidases. Therefore, these strains were used for lipase (EC 3.1.1.3) production by using used cooking and olive oils. Experimental planning for optimization of several parameters was carried out by a full factorial of two levels of three factors. Differential chromatographic behavior of lipase was investigated by immobilized 11 metal affinity chromatography (IMAC) by using epoxy-activated Sepharose 6B-imminodiacetic acid. One-step purification of lipase was devised by using a column of epoxy-activated Sepharose 6B-IDA-Cu (II). Purified lipase was immobilized by entrapment on calcium alginate beads and on microtiter plates in the presence of glutaraldehyde (GA). Therefore, lipase can be produced from these mutant strains by using olive oil in culture medium which can be purified by a one-step procedure by IMAC. The immobilized enzyme forms exhibited good storage and operational stabilities.
Keywords: Experimental Design; IMAC; Lipase Ec 3.1.1.3 Immobilization; Olive Oil; Wild-Type and Mutant Strains of Pseudomonas aeruginosa
Abbreviations: IMAC: Immobilized Metal Affinity Chromatography; IDA: Iminodiacetic Acid; GA: Glutaraldehyde; p-NPP: P-Nitrophenyl Palmitate; EPI: Epichlorohydrin; BSA: Bovine Serum Albumin
Introduction
Lipases (triacylglycerol acylhydrolase, C 3.1.1.3) are hydrolytic enzymes that catalyze the synthesis of glycerides in lipid-water interface. These enzymes also catalyze hydrolysis reactions, aminolysis, acidolysis (i.e exchange of an ester with an acid), alcoholysis (exchange of an ester to an alcohol), aminolysis, interesterification (exchange of an ester of the other ester) as well as esterification and transesterification reactions. Because of their stereoselective and regioselective properties by long chain fatty acids, lipases are widely used in pharmaceutical industry, detergents, cosmetics and textiles. They have also been used in dairy products in food industry as well as for biodiesel production (Ferrarezi, et al. [1,2]). There are a large number of microbial strains that have been used to produce lipase such as bacteria, molds and yeasts (i.e Bacillus spp., Pseudomonas spp., Staphylococcus spp., Aspergillus spp., Mucor spp., Penicillium spp., Candida spp. and Pichia spp (Christopher, et al. [3-5]). Lipase production depends on several factors, such as carbon sources, nitrogen sources, inducing agents and growth conditions of the microorganism such as aeration, pH and temperature (Christopher, et al. [3]). Inducers may be used to stimulate the production of lipase, such as triglycerides, free fatty acids, bile salts and hydrolysable esters (Gupta, et al. [6]). As far as literature search is concerned, there are several reports for extracellular lipase production from wild type strains of Pseudomonas aeruginosa by using appropriate culture media (Stuer, et al. [7,8]).
Although several lipases have been purified, there are no reports in the literature on the use of immobilized metal affinity chromatography (IMAC) for isolation of these enzymes by using a one-step procedure (Cheung, et al. [9]). Moreover, all purification procedures involved several precipitation and chromatographic steps which are time-consuming with low yields of lipase activity (Javed, et al. [5]). Microbial lipases are mostly extracellular exhibiting a molecular weight of 30-50 kDa with an optimal pH range of 7.5 - 9 and an optimum temperature of activity in the of range of 35-50 °C (Christopher, et al. [3]). Wild type and mutant strains of Pseudomonas aeruginosa 8602 have been widely used to produce altered aliphatic amidases of different specificities and affinities towards aliphatic amides (Brown, et al. [10,11]). Therefore, the aim of this work was to investigate the production of lipase from these wild-type and mutant strains by using suitable culture media as well as its purification by IMAC on a single step procedure. Subsequently, the enzyme will be immobilized in suitable supports for re-utilization purposes. To author´s knowledge, this is the first report on the use wild-type and mutant strains of Pseudomonas aeruginosa 8602 for lipase production by experimental design as well as purification by IMAC and immobilization on tissue culture microtiter plates.
Materials and Methods
Chemicals
Imidazole, iminodiacetic acid (IDA), Sepharose 6B, Triton X 100, Coomassie blue, glutaraldehyde (GA), p-nitrophenyl palmitate (p-NPP), sodium alginate and Arabic gum were obtained from Sigma -Aldrich (USA). Agar, peptone and yeast extract were supplied by Himedia, India. Olive oil was obtained from a local supermarket (Castelo de Moura – Extra Virgin oil, Lisbon) and used cooking oil was provided by ISEL canteen. Wild-type L10 and two mutant strains of Pseudomonas aeruginosa (3B and AI3) were kindly obtained from Dr. Paul Brown from King´s College London (UK).
Methods
Maintenance and Growth Conditions of Bacterial Strains in Solid Culture Media: Wild type L10 and mutant strains (3B and AI3) of Pseudomonas aeruginosa were cultured at 37ºC and maintained in two solid culture media at pH 7.2 as follows: culture medium A contained 2 g of agar, 1 ml of olive oil, 0.3 g of yeast extract, 0.1 g of peptone, 70 mg of K2HPO4, 30 mg of KH2PO4, 50 mg of MgSO4, 10 mg of MnCl2, 25 mg of (NH4)2SO4 and 10 mg of CaCl2 in a final volume of 100 ml of distilled water and culture medium B contained the same composition except olive oil was replaced by used cooking oil.
Lipase Production from Wild-Type and Mutant Strains in Liquid Culture Media: These mutant strains and two culture media (A and B) were used for lipase production. The liquid culture media A and B presented the same chemical composition as described above with the exception of agar. The strains were grown at 37ºC and 150 rpm in an incubator with orbital shaking for 4 days and samples were withdrawn at certain time intervals and centrifuged at 13000 rpm for one minute and the supernatant and pellet were stored at -20ºC.
Lipase Assay: Enzyme activity of lipase was measured by hydrolysis of p- nitrophenyl palmitate (p-NPP) producing p-nitrophenol and palmitic acid. p-NPP solution was prepared as follows: 6.229 mg of p-NPP, 1 ml of 2-propanol, 0.04 g of Triton X 100, 0.01 g of arabic gum in 10 ml of 20 mM Tris-HCl pH 8. The reaction mixture contained 80 μL of substrate solution and 20 μL of enzyme sample solution and the enzyme reaction was monitored at A415 in 96-well microtiter plate reader for an appropriate time interval. The molar extinction coefficient ((ε) of p-NPP was 18.100 M-1 cm-1. One enzyme unit (1U) was defined as the amount of enzyme required to produce 1 μmole of p-nitrophenol per minute under certain experimental conditions (Zivkovic, et al. [12]). Protein Assay: Protein assay was carried out by the Comassie blue dye binding method. These assays were performed in triplicates using bovine serum albumin (BSA) as the standard (Sedmark, et al. [13]).
Experimental Design for Optimization of Several Parameters of Lipase Production Conditions: The experimental design was arranged by a statistical factor of 23 with a total of eight experiments in duplicate. The screening experiment was designed to establish the effect of three factors: the growth temperature of bacterial strains (25ºC and 37ºC) as a numeric variable and wild-type L10 and mutant strain 3B of P. Aeruginosa strains and a suitable culture medium (olive oil and used cooking oil) as categorical variables. The measured response was the enzyme activity of lipase. The lower (-) and higher (+) levels were selected according to the results obtained from a preliminary study carried out earlier. The test matrix was developed, and the data obtained were introduced in the equation of the factorial model as follows:
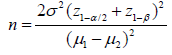
Where y is the lipase activity response variable, β0 is the mean of all responses, β is the regression coefficient and x are the prediction variable. The statistical analysis of the model was evaluated according to ANOVA methodology (Montgomery [14]) by using Design-Expert software version 9.0.3 (Shalabh [15]).
Preparation of Stationary Phases for IMAC: The activation of Sepharose 6B was carried out as described before (Martins, et al. [16]) by using epichlorohydrin (EPI) as the spacer arm and iminodiacetic acid (IDA) as the chelating agent.
Chromatographic Behavior of Lipases from Wild-Type and Mutant Strains of p. Aeruginosa on Immobilized Metal Chelates (IMAC): A rapid batch method carried out in ELISA microtiter plates was used to study the adsorption of lipase on stationary phases (Freixo, et al. [17]). In the present work, lipase adsorption to epoxyactivated Sepharose 6B- IDA–M(II) (S6) columns was considered to be partial when lipase activity was detected both at the washing and elution steps. On the other hand, lipase did not bind to the column when the total lipase activity applied to the column was recovered in five column volumes of buffer at the washing step, exhibiting a sharp activity peak. 96-well microtiter plates were used to study the adsorption of lipase from wild-type L10 and mutant strains 3B of P. Aeruginosa on immobilized metal chelates of Cu(II), Ni(II), Zn(II) and Co(II). The culture supernatant containing lipase activity (specific activity 0,009 U/mg protein) was diluted in 20 mM sodium phosphate buffer containing 1M NaCl at the corresponding pH. Three pH values (6, 7 and 8) were tested to investigate the best conditions that promote adsorption of the enzyme to immobilized metal chelates. Desorption of the enzyme from metal chelates was carried out by using 20 mM phosphate buffer containing 1 M NaCl and 500 mM imidazole. All washing and elution fractions were assayed for lipase activity and these experiments were performed in triplicates at room temperature.
Purification of Lipase from Mutant Strains of P. Aeruginosa by IMAC: According to the results of the chromatographic behavior performed on 96-well microtiter plates, a set of experimental conditions were selected for purification of lipase from mutant strains of P. Aeruginosa. A column containing 5 mL of sedimented epoxy-activated Sepharose 6B-IDA (S6) as the stationary phase was washed sequentially with 5 volumes of 50 mM copper sulfate, water and then finally with 20 mL of equilibration buffer at the corresponding pH. The culture supernatant containing lipase activity was diluted 1:2 with equilibration buffer and applied to the column at a flow rate of 15 mL/h. The column was then washed with equilibration buffer and the chromatographic fractions were collected and read at A280. Once the A280 was less than 0.05, the enzyme was desorbed from the column with a linear gradient of imidazole (0-500 mM) in the same buffer system at a flow rate of 30 mL/h and fractions were assayed for enzyme activity and protein content.
Electrophoretic Analysis of Lipase from Mutant Strains of Pseudomonas Aeruginosa: SDS-PAGE and native PAGE of purified lipase and culture supernatants were carried out as mentioned previously (Bloom, et al. [18]) and stained for protein with silver nitrate.
Lipase Immobilization: The immobilization of lipase was performed by entrapment on calcium alginate beads as well as by covalent method using GA on 96 and 24-well tissue culture microtiter plates.
Lipase Entrapment in Calcium Alginate Beads: A solution of 4% (wt/v) of sodium alginate and a 2% (w/v) solution of CaCl2 were prepared previously and sodium alginate solution (2 ml) was mixed with 2 ml of purified enzyme (specific activity 0,2 U/ mg protein) from mutant strain 3B of P. Aeruginosa. An automatic micropipette was used to make small drops which were dripped into CaCl2 solution and spherical beads of different diameters were obtained. Calcium chloride solution was gently stirred for 1 h at room temperature and the calcium alginate beads containing lipase were recovered by filtration of CaCl2 solution. These beads containing immobilized enzyme were washed with 25 mM phosphate buffer at pH 8 and air dried at room temperature. The bead size was measured by using an optical microscope equipped with a digital image analyzer which was measured at several angles and mean values were obtained.
The recovery of immobilized enzyme activity was determined in triplicates by measuring the enzyme activity of the free and immobilized forms of lipase by ELISA reader at A415. The immobilized form of lipase activity was measured by using calcium alginate beads in a reaction mixture containing 100 μL of substrate solution which was incubated at room temperature for 10 min. Subsequently, the supernatants were read at A415 and compared with the control samples. The enzyme beads were stored in 25 mM phosphate buffer at pH 8.0 at 4ºC. The storage stability of immobilized enzyme was carried out in 25 mM phosphate buffer pH 8.0 at 4ºC. The beads were used to assay for enzyme activity three times a week for one month by using the methodology described above. The operational stability was carried out to determine reutilization of the entrapped lipase which was assayed with p-NPP as described above. Ten cycles were performed in 10 days and each cycle of the immobilized lipase was run for 45 min. The residual enzyme activity was expressed as the percentage of the activity at several cycles compared with the first cycle.
Immobilization of Lipase with Glutaraldehyde on Microtiter Plates: 96 and 24-well microtiter plates were used as supports for lipase immobilization as described previously (Charneca, et al. [19]). As far as 96-well microtiter plates are concerned, 0, 10, 20, 40, 60, 80 and 100 μL of purified lipase (0,2 U/ mg protein) from mutant strain 3B of P. Aeruginosa were added to the wells and the solution allowed to dry at room temperature. Subsequently, 25% (v/v) of (GA) were added at different concentrations (0, 2, 4, 6, 10 and 15% GA) and allowed to dry. The wells were washed three times with 0.1 M phosphate buffer at pH 8 and the immobilized enzyme was stored at 4ºC in 25 mM phosphate buffer at pH 8.0. Lipase assay was carried out in triplicates to measure the immobilized lipase activity by adding 100 μL of substrate solution in microtiter plates wells which were incubated for 10 min at room temperature. The supernatant was read at A415 in ELISA plate reader in the test wells as well as in the control wells. 24-well microtiter plates were used to immobilize lipase by adding to the wells 0, 50, 100, 200, 300, 400 and 500 μL of purified enzyme (0,2 U/ mg protein) from 3B mutant strain which were allowed to dry at room temperature.
After the enzyme solution had dried, 25% (v/v) GA was added at different concentrations (0, 2, 4, 6 and 8% GA) and allowed to dry at room temperature. The wells were washed three times with 0.1M phosphate buffer at pH 8 and lipase assay was carried out in triplicates to measure the activity of immobilized enzyme by adding 150 μL of substrate solution to the wells which were incubated for 10 min at room temperature. The supernatant was read at A415 in ELISA plate reader in the test wells as well as in the control wells. The immobilized lipase in microtiter plates were kept in 25 mM phosphate buffer pH 8.0 at 4ºC for storage stability purposes which was carried out as described for calcium alginate beads. The operational stability was also performed as described for enzyme beads.
Results and Discussion
Lipase Production from Wild-Type and Mutant Strains of P. Aeruginosa
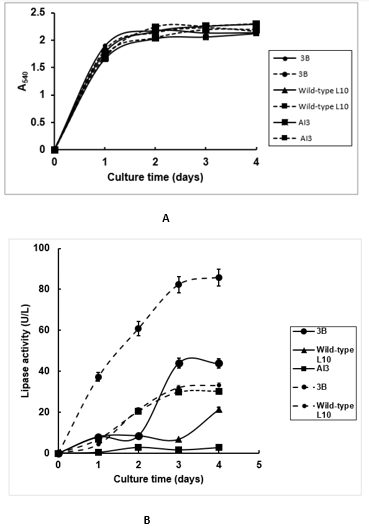
Figure 1: A. Lipase production from wild-type (L10) and mutant strains of Pseudomonas aeruginosa in two culture media. Growth curves of wild-type L10 and mutant strains (3B and AI3) of P. aeruginosa in two culture media. Culture medium A with olive oil (- - - -) and culture medium B with used cooking oil ( ______).
B. Lipase production from wild-type (L10) and mutant strains of Pseudomonas aeruginosa in two culture media. Lipase production from wild-type and mutant strains in both culture media. Culture medium A with olive oil (- - - -) and culture medium B with used cooking oil ( ___).
Lipase production from wild-type L10 and mutant strains AI3 and 3B of P. Aeruginosa was investigated by using two culture media as shown in (Figure 1). The growth culture in terms of biomass was similar for all mutant strains in both culture media (Figure 1). However, lipase activity was higher in culture medium containing olive oil compared with the used cooking oil (85,9 and 44,5 U/L), respectively (Figure 1B). The highest lipase activity was observed in both media for mutant strain 3B followed by wild-type L10 and AI3 strains as revealed in (Figure 1B). As far as literature is concerned, there are two reports about the use of olive oil for lipase production from Pseudomonas aeruginosa strains (760 and 41600 U/L). However, it is difficult to compare lipase levels described in the literature because the assay method as well as assay conditions are different from the present work (Mobarak Qamsari, et al. [20,21]). To author´s knowledge, there are no reports in the literature about the use of used cooking oil for lipase production from wild-type and mutant strains of P. Aeruginosa.
Experimental Planning for Optimization of Several Parameters for Lipase Production
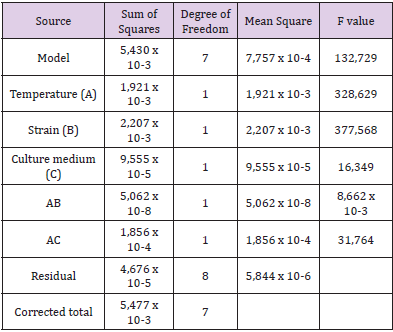
Table 1: Analysis of variance (ANOVA) of the factorial design model proposed.
Note: a. Test distribution is Normal.
A preliminary optimization of some key factors affecting lipase production was carried out above. Subsequently, experimental planning was performed on a statistical factorial of 23 with a total of eight experiments in duplicate. This study was carried out to establish the effect of three factors as follows: the growth temperature of microbial strains (25ºC and 37ºC), the nature of strain of P. Aeruginosa (Wild-type L10 and 3B) and the appropriate culture medium (olive oil and used cooking oil). The statistical factorial design is a tool that provides the minimum number of experiments required to evaluate the simultaneous effects of multiple variables and multi-variables interactions on predetermined processing objective (Montgomery, et al. [14,15]). The summary of the analysis of the variables of the factorial model are presented in (Table 1) which exhibit the average responses and the predicted results. The effects of each variable and multivariable interaction and predetermined process objectives were obtained by the use of Design-Expert software version 9.0.3.
The high value of F model (132.729) implies that the model is significant and there is a small percentage of this high value F occurs due to noise. An empirical equation was obtained to estimate the value of the response (slope/sensitivity): y=0.04201+0.02191 A-0.02349 B+0.00489 C+0.00011 AB-0.00681 AC Where y is the enzyme activity of lipase and A, B and C are the values of temperature, bacterial strain and culture medium, respectively. This equation is used to determine an optimum response. The data in (Figure 2A) revealed the three-dimensional representation for the interaction of temperature parameter (25ºC and 37ºC) and the culture medium (olive oil and used cooking oil) on lipase activity from mutant strain 3B of P. Aeruginosa. The optimized conditions consisted of a temperature of 37ºC and a culture medium with olive oil since (Figure 2A) displayed highest enzyme activity for these values. The data in (Figure 2B) revealed the three-dimensional representation for interaction parameters of strain (Wild-type L10 and Ph3B) and temperature (25 ºC and 37ºC) on lipase activity from P. Aeruginosa in the culture medium with olive oil. The optimized conditions consisted of a temperature of 37ºC with mutant strain 3B of P. Aeruginosa, since these parameters exhibited the highest enzyme activity. As far as literature is concerned, there are no reports about experimental planning for lipase production from P. Aeruginosa.
Figure 2:
A. Experimental planning for optimization of several parameters for lipase production. Three-dimensional representation for interaction of temperature parameters and culture medium on lipase activity from mutant strain 3B of P. aeruginosa.
B. Experimental planning for optimization of several parameters for lipase production. Three-dimensional representation for interaction parameters of mutant strain and temperature on lipase activity from P. aeruginosa in culture medium with olive oil.
Chromatographic Behavior of Lipases from Wild-Type and Mutant Strains of p. Aeruginosa on Immobilized Metal Chelates
The chromatographic behaviour of lipases is summarized in (Table 2A), for epoxy-activated Sepharose 6B-IDA (S6) as the stationary phase as well as for different immobilized metal chelates. The results from adsorption assays strongly suggest that immobilized Cu (II) chelates of this stationary phase exhibited the highest affinity for both lipases (Table 2A). Moreover, all immobilized metal chelates of epoxy-activated Sepharose 6B-IDA adsorbed both lipases at pH 6.0 and 8.0 whereas the adsorption was rather weak at pH 7.0. X-ray structure of lipase from P. Aeruginosa has been reported in the literature which exhibits a monomer with a single polypeptide chain. It contains seven histidine (His) residues per enzyme molecule at positions 14, 81, 83, 109, 238, 251 and 276 of the polypeptide chain (Nardini, et al. [22]). Lipase adsorption observed in IMAC is essentially governed by the spatial distribution of histidine residues on the protein surface. Therefore, the differential chromatographic behaviour of lipase from wildtype and mutant strains is due to the presence of different number of histidine residues on their surfaces. It is important to point out that IMAC was also successfully used for lipase purification from basidiomycete strains such as Pleurotus ostreatus and Ganoderma lucidum (data not shown). The culture supernatant containing lipase activity from mutant strain AI3 was purified in a single step by IMAC on epoxy-activated Sepharose 6B-IDA-Cu(II) at pH 8.0 with a specific activity of about 0,2 U/mg protein, a recovery of about 225% and purification factor of about 20 (Table 2B). Based on the data obtained from the qualitative and quantitative adsorption tests, several chromatographic runs were carried out on columns packed with 5 ml of sedimented stationary phase which was charged with transition metal ions at different pH values.
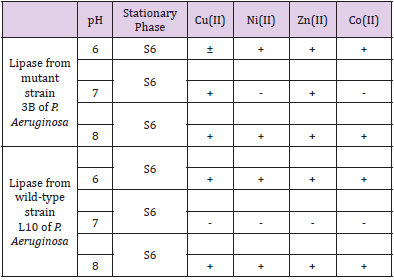
Table 2A: Chromatographic behavior of lipase from wild-type L10 and mutant strain 3B of P. aeruginosa on immobilized metal chelates of epoxy activated Sepharose 6B-IDA (S6).
Note:
1. +, denotes that enzyme activity adsorbed to the stationary phase.
2. ±, denotes partial binding since enzyme activity was detected both at the washing and elution steps.
-, denotes that enzyme activity did not bind to stationary phase since total enzyme activity was recovered at the washing step.
All chromatographic runs were carried out in triplicates.
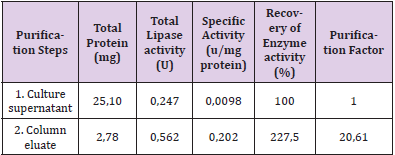
Table 2B: Purification of lipase from a mutant strain AI3 of P.aeruginosa by IMAC on epoxy activated Sepharose 6B-IDACu( II) at pH 8.0.
The concentration of imidazole required for protein desorption from the column was found to increase as a function of the pH of the mobile phase as predicted by IMAC theory. For instance, according to (Figures 3A & 3B), it was found that the concentration of imidazole required for the desorption of lipase from the column packed with epoxy-activated Sepharose 6B- IDA-Cu(II) increased as a function of the pH of the mobile phase. Several research workers have observed the same effect in retention of peptides and proteins lacking histidyl groups on immobilized metal chelates which may be due presumably to the binding of unprotonated alpha-amino group to metal chelates at higher pH (Porath, et al. [23,24]). By lowering the pH of the mobile phase to 6, selective interactions occur with the available histidine residues on the surface of the protein, and thus, the desorption of the protein is achieved with a lower imidazole concentration in the mobile phase (Martins, et al. [16]). In order to confirm that the interaction of lipases with immobilized Cu(II) chelates was due to the coordination between available histidine residues on enzyme molecules and chelated transition metal ions, chromatographic runs were carried out with imidazole in the equilibration buffer. The results revealed that lipase did not adsorb to immobilized Cu(II) chelates in the presence of 20 mM imidazole in the equilibration buffer (Figure 3C). It was also observed that lipase did not adsorb to epoxy-activated Sepharose 6B-IDA column without Cu ions which suggests that adsorption of lipase on this affinity matrix was apparently due to the specific interaction of immobilized metal chelates and histidine residues available on lipase (data not shown).
The recovery of lipase activity was higher that 100% (Table 2B) and this result may be due to either the presence of small molecules as inhibitor in the culture supernatant or a change in conformation on the purified enzyme molecule. Moreover, published work has been reported that lipase was found in culture supernatant of P. Aeruginosa as aggregates of high molecular weight complexes with lipopolysaccharides (Stuer, et al. [7]) which could apparently reduce its enzyme activity. The purified enzyme was apparently homogenous on SDS and native PAGE running as a single protein band with a Mr of 42 KD (data not shown). As far as published reports are concerned in the literature, lipases have been purified by using several time-consuming purification steps with low final recoveries of enzyme activity of about 7 and 15% and purification factor of 98 and 1000, respectively (Borkar, et al. [21,7]). Regarding the specific activity of purified lipase in the literature, it is difficult to compare with the present data since different assay methods, assay conditions and enzyme units were used in their work respectively (Borkar, et al. [21,7]). As far as the literature is concerned, the main advance in the present work involves the differential chromatographic behaviour of lipase on stationary phases of IMAC by using several immobilized metals chelates. On the other hand, a one-step purification of lipase from mutant strains of P. Aeruginosa by IMAC is a novel procedure. This chromatographic technique was also successfully used for lipase purification from basidiomycete strains (data not shown).
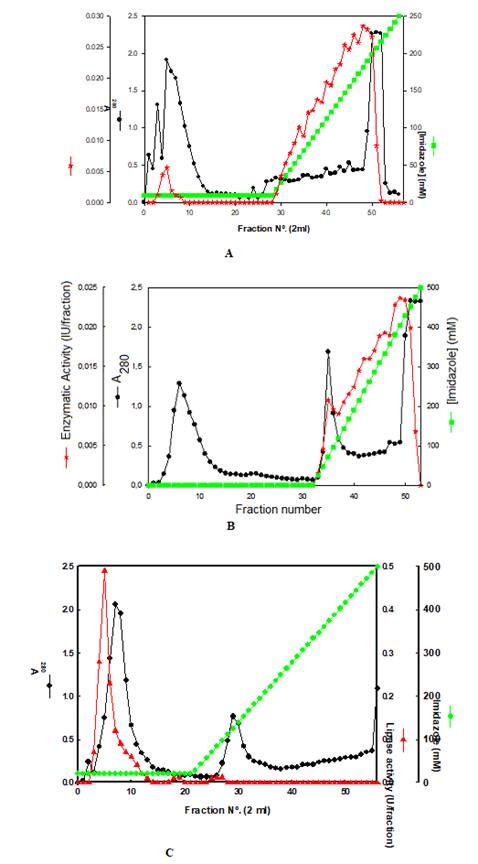
Figure 3:
A. Purification of lipase from mutant strains of P. aeruginosa on epoxy—activated Sepharose 6B-IDA-Cu (II) column. A – Mutant strain 3B with a linear gradient of 0 to 250 mM of imidazole at pH 7.0
B. Purification of lipase from mutant strains of P. aeruginosa on epoxy—activated Sepharose 6B-IDA-Cu (II) column. Mutant strain AI3 with a linear gradient of 0 to 500 mM of imidazole at pH 8.0
C. Purification of lipase from mutant strains of P. aeruginosa on epoxy—activated Sepharose 6B-IDA-Cu (II) column. Mutant strain AI3 with an equilibration buffer with 20 mM imidazole and a linear gradient of 20 to 500 mM of imidazole at pH 8.0 on epoxy—activated Sepharose 6B-IDA-Cu (II) column.
Lipase Immobilization
Lipase was immobilized by entrapment on calcium alginate beads and covalent immobilization with glutaraldehyde on microtiter plates. Preliminary experiments revealed that beads of high diameter exhibited low lipase activity. Therefore, we investigated the effect of bead diameter on immobilized lipase activity. The data presented in (Figure 4A) revealed that optimum lipase activity was observed for beads with diameter range of 1 to 2 mm. These results may be explained due to external and internal mass transfer limitations for immobilized enzymes. The recovery of immobilized enzyme on calcium alginate beads was about 40% compared with the free enzyme (Figure 4B). The storage stability of immobilized lipase was investigated by measuring residual enzyme activity of calcium alginate beads which revealed about 15% loss of enzyme activity (data not shown). On the other hand, the operational stability of immobilized lipase was investigated for 10 cycles which revealed a t1/2 of 41,75 cycles or days (Figure 4B). As far as literature search is concerned, there is only one published work on immobilization of lipase from P. Aeruginosa on calcium alginate beads which reported immobilization yield of about 93% (Kavardi, et al. [25]). On the other hand, lipase from Corynebacter sp. was also immobilized on calcium alginate beads which exhibited about 87 and 68% of original enzyme activity for 21 and 40 days of storage stability at 4ºC, respectively (Roy, et al. [26]).
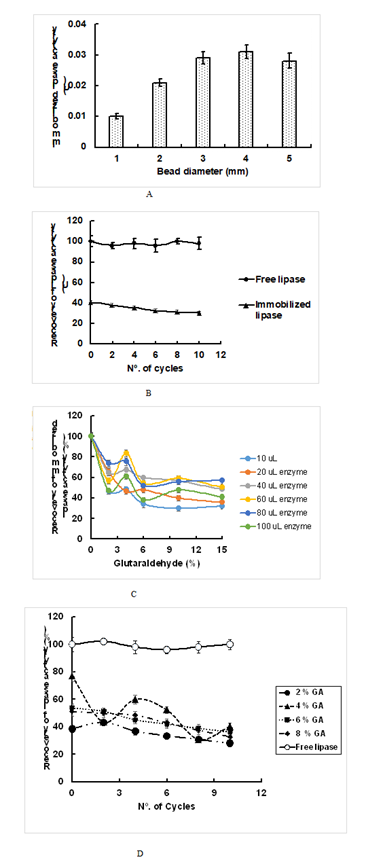
Figure 4:
A. Lipase immobilization from mutant strain 3B of Pseudomonas aeruginosa. Entrapment on calcium alginate beads. Effect of bead diameter on biocatalysis of immobilized lipase
B. immobilization from mutant strain 3B of Pseudomonas aeruginos. Entrapment on calcium alginate beads. Operational stability of immobilized lipase on calcium alginate beads for 10 cycles of 45 min.
C. Lipase immobilization from mutant strain 3B of Pseudomonas aeruginosa. Covalent binding on microtiter plates by using glutaraldehyde. Effect of glutaraldehyde concentration on recovery of immobilized enzyme activity in 96-well microtiter plates.
D. Lipase immobilization from mutant strain 3B of Pseudomonas aeruginosa. Covalent binding on microtiter plates by using glutaraldehyde. Operational stability of immobilized lipase from mutant strain 3B on 24-well microtiter plates by using 200 uL of purified enzyme (0.54U/mL).
However, none of these reports provide information about the re-utilization of immobilized lipase as well as its operational stability. Covalent immobilization of lipase was carried out in 96 and 24 -well microtiter plates. The effect of glutaraldehyde (GA) concentration on immobilized lipase activity was investigated on 96-well microtiter plates by using different volumes of purified enzyme. The data revealed in (Figure 4C) strongly suggest that high concentrations of GA significantly decreased the enzyme activity since this bifunctional reagent is highly toxic for proteins by damaging their tertiary structure. On the other hand, high amounts of enzyme used for covalent immobilization revealed high enzyme activity for most concentrations of GA (Figure 4C). The storage stability of immobilized lipase was investigated which revealed about 25% loss of enzyme activity (data not shown). On the other hand, the operational stability of immobilized lipase on 24 -well microtiter plates were investigated for 10 cycles which revealed t1/2 of 27.8, 7,3, 20,8 and 13,2 cycles or days for 2, 4, 6 and 8% GA, respectively (Figure 4D). Regarding the literature, there are no reports about lipase immobilization on microtiter plates in the presence of GA. Lipase immobilization on microtiter plates was a novel procedure which exhibited high enzyme recovery, storage and operational stabilities. However, some published work reported the immobilization of other enzymes, whole cells and cell-free extract of microtiter plates by using GA (Charneca, et al. [19,27,28]). These reports investigated the storage stability of immobilized glucose nanosensors and glucose oxidase which was 7 and 4 months and operational stability of at least 20 and 30 cycles, respectively (Charneca, et al. [19,28]).
Acknowledgement
“This work was supported by: European Investment Funds by FEDER/COMPETE/POCI– Operacional Competitiveness and Internacionalization Programme, under Project POCI-01- 0145-FEDER-006958 and National Funds by FCT - Portuguese Foundation for Science and Technology, under the project UID/ AGR/04033/2013”.
References
- Ferrarezi A, Ohe T, Borges J, Brito R, Siqueira M, et al. (2014) Production and characterization of lipases and immobilization of whole cell of the thermophilic Thermomucor indicae seudaticae N31 for transesterification reaction. J of Molecular Catalysis B Enzymatic 107: 106-113.
- Anobom CD, Pinheiro AS, De Andrade RA, Aguieiras ECG, Andrade GC, et al. (2014) Review Article from Structure to Catalysis: Recent Developments in the Biotechnological Applications of Lipases. BioMed Research International 68450: 1-11.
- Christopher L, Kumar H, Zambare VP (2014) Enzymatic biodiesel: Challenges and opportunities. Applied Energy 119: 497-520.
- Sharma R, Chisti Y, Banerjee U (2001) Production, purification, characterization, and applications of lipases. Biotechnology Advances 19(8): 627-662.
- Javed S, Azeem F, Hussain S, Rasul I, Siddique MH, et al. (2017) Bacterial lipases: A review on purification and characterization. Progress in. Biophysics and Molecular Biology 132: 23-34.
- Gupta R, Gupta N, Rathi P (2004) Bacterial lipases: an overview of production, purification and biochemical properties. Applied Microbiology and Biotechnology 64(6): 763-781.
- Stuer W, Jaeger KE, Winkler UK (1986) Purification of Extracellular Lipase from Pseudomonas aeruginosa. J Bacteriology 168(3): 1070-1074.
- Jaeger KE, Ransac S, Dijkstra BW, Colson C, Heuvel MV, et al. (1994) FEMS Microbiology Reviews 15: 29-63.
- Cheung RC, Wong JH, Ng TB (2021) Immobilized metal ion affinity chromatography: a review on its applications. Appl Microbiol Biotechnol 96(6): 1411-1420.
- Brown PR, Clarke PH (1972) Amino acid substitution in an amidase produced by an acetanilide-utilizing mutant of Pseudomonas aeruginosa. J Gen Microbiol 70(2): 287-298.
- Andrade J, Karmali A, Carrondo MA, Frazão C (2007) Structure of amidase from Pseudomonas aeruginosa showing a trapped acyl transfer reaction intermediate state. J Biol Chem 282(27): 19598-19605.
- Zivkovic L, Zivkovic L, Babic B, Kokunesoski M, Jovic B, et al. (2015) Immobilization of Candida rugosa lipase by adsorption onto biosafe meso/macroporous silica and zirconia. Biochemical Engineering Journal 93: 73-83.
- Sedmak JJ, Grossberg SE (1977) A rapid, sensitive, and versatile assay for protein using Coomassie Brilliant Blue G-250. Anal Biochem 79(1-2): 544-552.
- Montgomery DC (2009) Factorial and fractional factorial experiments for process design and improvement. Pp. 549-601. In: Introduction to Statistical Quality Control (3rd)., John Wiley & Sons, Inc. Jefferson City, MO, USA.
- Shalabh (2009) Experimental design in biotechnology and 2k factorial design. In: Statistical Analysis of Designed Experiments,3rd ed. Springer, New York, pp 287-290.
- Martins S, Karmali A, Serralheiro ML (2011) Chromatographic behaviour of monoclonal antibodies against wild-type amidase from Pseudomonas aeruginosa on immobilized metal chelates. Biomed Chromatogr 25(12): 1327-1337.
- Freixo MR, Karmali A, Arteiro JM (2008) Production of polygalacturonase from Coriolus versicolor grown on tomato pomace and its chromatographic behaviour on immobilized metal chelates. J Ind Microbiol Biotechnol 35(6): 475-484.
- Bloom H, Beier H, Gross HS (1987) Improved silver staining of plant proteins, RNA and DNA in polyacrylamide gels. Electrophoresis 8(2): 93-99.
- Charneca A, Karmali A, Vieira M (2015) Non-enzymatic assay for glucose by using immobilized whole-cells of E. coli containing glucose binding protein fused to fluorescent proteins. Sensors and Actuators B Chemical 221: 236-241.
- Mobarak Qamsari E, Kasra Kermanshahi R, Moosavi nejad Z (2011) Isolation and identification of a novel, lipase-producing bacterium, Pseudomnas aeruginosa KM110. Iran J Microbiol 3(2): 92-98.
- Borkar PS, Bodade RG, Rao SR, Khobragade CN (2009) Purification and characterization of extracellular lipase from a new strain Pseudomonas aeruginosa SRT 9. Brazilian J of Microbiology 40: 358-366.
- Nardini M, Lang DA, Liebeton K, Jaeger KE, Dijkstra BW (2000) Crystal Structure of Pseudomonas aeruginosa Lipase in the Open Conformation. The prototype for family i.1 of bacterial lipases. J Biol Chem 275(40): 31219-31225.
- Porath J (1992) Immobilized metal ion affinity chromatography. Protein Expr Purif 3(4): 263-281.
- Todd RJ, Johnson D, Arnold FH (1994) Multiple site binding interactions in metal-affinity chromatography. I Equilibrium binding of engineered histidine containing cytochromes c. J Chromatogr A 662(1): 13-26.
- Kavardi SSS, Alemzadeh I, Kazemi A (2012) Optimization of lipase immobilization. IJE Transactions C: Aspects 25(1): 1-10.
- Roy N, Ray L, Chattopadhyay P (2004) Production of lipase in a fermentor using a mutant strain of Corynebacterium species: Its partial purification and immobilization. Indian Journal of Experimental Biology 42(2): 202-207.
- Brandt B, Hidalgo A, Bornscheuer UT (2006) Immobilization of enzymes in microtiter plate scale. Biotechnology J 1(5): 582-587.
- Ukeda H, Fujita Y, Ohira M, Sawamuraj M (1996) Immobilized Enzyme-Based Microtiter Plate Assay for Glucose in Foods. Agric Food Chem 44(12): 3858-3863.
