 Research Article
Research ArticleABSTRACT
The viral nucleocapsid (N) protein plays a vital role in the replication and transcription of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) genome. In the current study pharmacophore-based database screening, molecular docking analysis, toxicity prediction and molecular dynamics simulation were used to identify potential novel Nucleocapsid (N) protein inhibitors. Pharmacophore screening performed using Zinc database resulted in the identification of four prospective compounds viz., ZINC55083699, ZINC12487932, ZINC81013764, and ZINC68104820 as potential N-capsid inhibitor candidates with desired pharmacokinetic properties. All the 4 compounds when subjected to molecular dynamics simulation at a 100ns time scale exhibited a stable configuration in the complete solvent system. The result indicates that all the four potential compounds are worth exploring as inhibitors of SARS-CoV-2 Nucleocapsid (N) protein.
Keywords: SARS-CoV-2; Covid-19, Nucleocapsid (N) Protein; Zinc Database; Pharmacophore Modelling; Virtual Screening; Molecular Docking; Molecular Dynamics Simulation; Nucleocapsid Protein Inhibitors; Drug Identification
Abbreviations: HTVS: High-Throughput Virtual Screening; SP: Standard Precision; XP: Extra Precision; MD: Molecular Dynamics; RMSF: Root-Mean-Square Fluctuations; CADD: Computer-Aided Drug Design
Introduction
The current severe acute respiratory syndrome pandemic caused by SARS-COV-2 infection is a serious global health concern due to its rapid spread over with high mortality [1]. According to WHO, globally COVID-19 has spread to more than 224 countries with 273,538,415 million confirmed cases and 5,356,949 million deaths till 17th December, 2021 [2]. COVID-19 spreads through droplets, aerosols, feces, and oral mucus membranes of infected persons. The main symptoms are typical acute respiratory infection, cough, fever, myalgia, pharyngeal erythema, sneezing, acute kidney injury, fatigue, acute liver injury, diarrhea, sore throat, breathing impairment, acute renal failure, encephalopathy, irregular coagulation, and vomiting [3,4]. Based on serology and heritability, corona viruses are classified into four types: α, β, γ, and δ [5]. While α and β corona viruses predominantly infect mammals whereas, γ and δ corona viruses primarily infect birds. So far, seven different varieties of human corona viruses were documented including HCoV-NL63 and HCoV-229E that belongs to α corona virus, and HCoV-OC43, HCoVHKU1, SARS-CoV, MERS-CoV, SARS-CoV-2 to the β corona virus [1].
SARS-CoV-2 belongs to the β coronavirus type, with a singlestranded RNA genome similar to MERS-CoV and SARS-CoV. The new βCoV exhibits 88% sequence similarity with two bat-derived SARSlike coronaviruses. The first open reading frame (ORF) represents roughly 67% of the entire genome encoding 16 non-structural proteins. Coronaviruses have numerous structural proteins that are highly identical across their host species including the small envelope proteins (E), the trimeric spike protein (S), matrix glycoprotein (M) and Nucleocapsid (N) protein in the interior of the virus [6]. A few coronaviruses contain a third membrane-bound glycoprotein, HE (hemagglutinin-esterase). The Nucleocapsid (N) protein comprises a copious part of structural proteins in corona viruses. During replication and transcription, it provides vital information for helical ribonucleoproteins in the infected cells during viral RNA synthesis. Furthermore, the Nucleocapsid (N) protein contains three domains: an RNA-binding domain at the N-terminal region, an essentially tangled central SER/ARG-rich linker that might have the protein’s primary sites of phosphorylation, and a C-terminal dimerization domain. Disruption of the communication between RNA and viral Nucleocapsid (N) protein can block the replication of the viral genome [7,8]. Therefore, Nucleocapsid (N) protein seems to be a good drug target. At present, several vaccines are at various stages of development to combat the spread of COVID-19 viz., NVXCoV2373, PittCoVacc,
Triple Antigen Vaccine, Ad5-nCoV, Coroflu, LV-SMENP-DC, ChAdOx1, mRNA-1273, BNT162b1 and INO-4800 [9-15]. In view of rapid mutations occurring in SARS-CoV-2 genome and development of several new potent variants (Alpha, Beta, Gamma, Delta, and Omicron) there is an urgent need to find effective drugs/ vaccines that can stop the SARS-CoV-2 viral multiplication. Using computational tools, several research groups have attempted to find potential drugs for the inhibition of COVID-19 virus. In the current study, computer-aided drug design (CADD) and pharmacophore-based virtual screening was used to screen novel inhibitors targeting Nucleocapsid(N)protein of SARS-CoV-2 from the Zinc database.
Materials & Methods
E-Pharmacophore Modeling and Molecular Docking
The 3D coordinates of Nucleocapsid (N) protein from SARSCoV- 2 (PDB ID: 6M3M) was retrieved [16] from PDB and preprocessed using Protein Preparation-wizard of Schrodinger version 5.5. During pre-processing, bond orders were assigned, unwanted water molecules were eliminated, formal charges and hydrogen atoms were added and all atom force field charges (OPLS3) were assigned to Nucleocapsid (N) protein. Receptor energy minimization was terminated when the energy converged or when the root mean square deviation reached a maximum cutoff of 0.30 Å [17,18]. In the structure based docking, crucial binding pocket residues of the Nucleocapsid (N) protein viz., THR 55, ALA 56, ARG 89, TYR 110, TYR 112 were utilized for grid box generation [16]. In the ligand preparation (i) OPLS-2005 force field was used, all conceivable ionization states at pH 7.0±2.0 were added, the desalt preference were created, tautomers were generated for all conformers. E-Pharmacophore mapping was performed using the scoring function against atom centers. The Glide XP conformers were specified as input to construct pharmacophore sites. The robust hypothesis remained subsequently utilized as a query for the database survey to identify potential inhibitors. A set of six innate chemical features, namely hydrogen bond acceptor (A), hydrogen bond donor (D), hydrophobic group (H), negative ionizable region (N), positive ionizable region (P), and aromatic ring (R) were used for pharmacophore construction.
E-Pharmacophore Generation
For E-pharmacophore modelling, Nucleocapsid (N) protein inhibitors were selected based on the 3D structure of target protein (PDB ID: 6M3M) from protein databank. The prepared ligand and protein files were imported into Glide XP of Schrodinger software for docking analysis and the best conformer was subsequently used for E-pharmacophore construction. Schrödinger E-pharmacophore module was used to construct pharmacophore hypothesis. The constructed pharmacophore model comprises three-point pharmacophore hypotheses covering two hydrogen bond donors D3 & D4 and one hydrogen bond acceptor A1. The distances between pharmacophore sites A1 and D3, A1 and D4, and D3 and D4 were 2.452, 8.016, and 6.734, respectively. The pharmacophore site D3 was found in the cyclic amine NH group, acceptor A1 was found in the cyclic keto carbonyl CO group, and donor D4 was found in the side chain amine NH group of pj34. The overall screening flowchart (Figure 1), 2D structure of the reference ligand pj34 (Figure 2) and the distances and angles between the pharmacophore and their sites (Figures 3 & 4) were depicted below.
Chemical Warehouse Screening Based on Pharmacophore Features
Pharmacophore-based database screening is a rapid, accurate computational technique used in drug discovery to predict hit compounds with desirable pharmacokinetic properties [19]. Lipinski rules were applied to eliminate non-drug-like compounds; therefore, drugs with a molecular weight below 500 Da, a partition coefficient logP < 5, less than 5 hydrogen bond donors, less than 10 hydrogen bond acceptors were excluded. The ligands with suitable pharmacokinetic properties [20] were utilized to identify potential inhibitors of SARS-CoV-2 Nucleocapsid (N) protein. All the docking studies were carried out using Glide module. The Glide docking procedure involves three significant filtering phases: highthroughput virtual screening (HTVS), standard precision (SP), and extra precision (XP). Each docking mode, from HTVS to SP and SP to XP involves a Glide score, which represents the binding affinity used for ranking the ligands. A total of 4000 compounds screened from Zinc libraries based on the pharmacophore features were docked into the active site of SARS-CoV-2 Nucleocapsid (N) protein. Validated 3D pharmacophore models (ADD) of SARS-CoV-2 Nucleocapsid (N) protein inhibitors were used for the identification of competent molecules as well as coveted trait from large chemical warehouse i.e., Zinc database accompanied by fitness grade ranges >1 and 1000 hits were obtained based on the fitness score and pharmacophore matches.
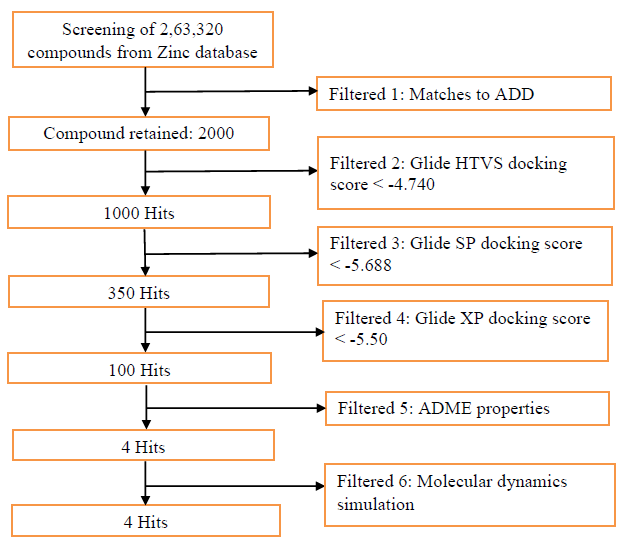
Figure 1: Flowchart of methodology employed during insilico screening for identification of novel SARS CoVID-19 Nucleocapsid (N) protein inhibitors.
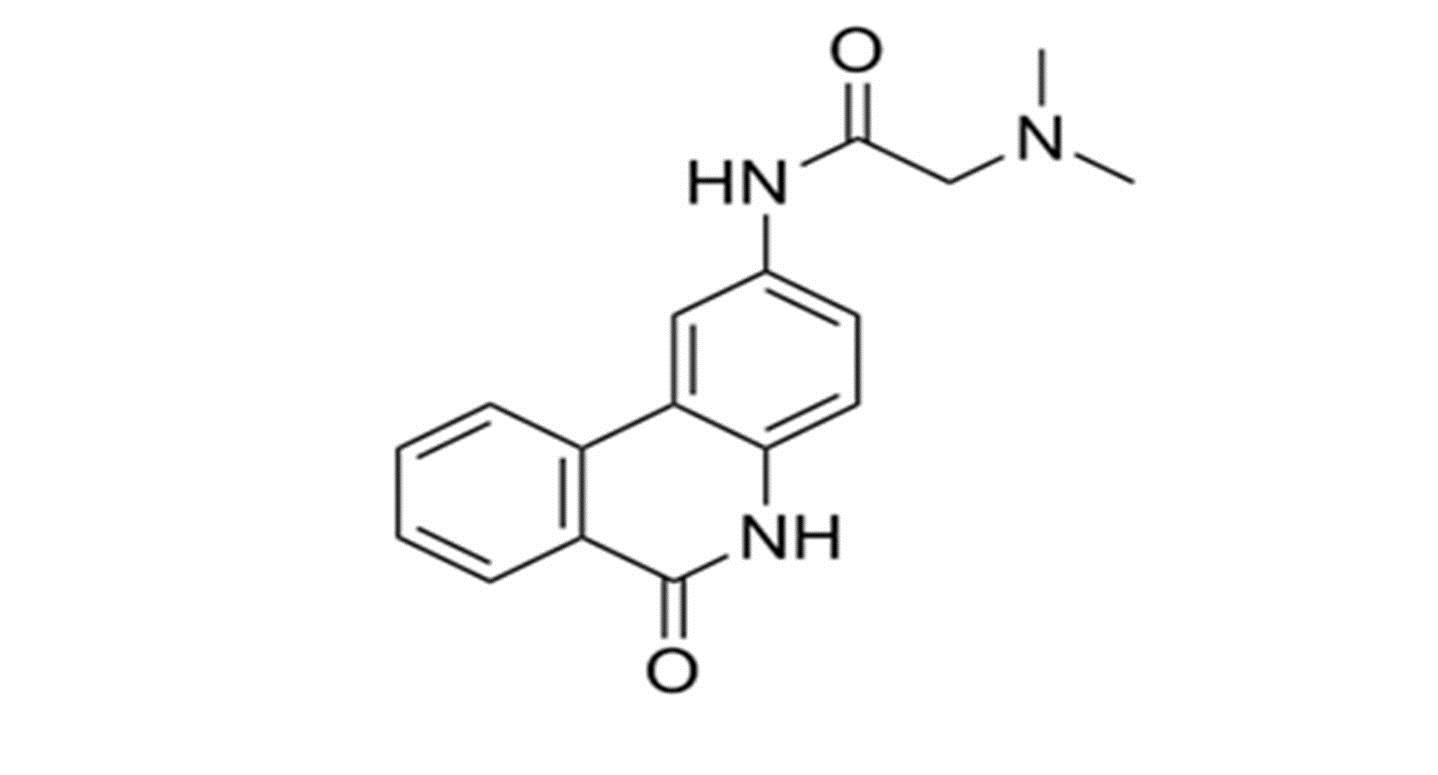
Figure 2: The 2D structures of the SARA-CoV-2 Nucleocapsid (N) protein inhibitor.
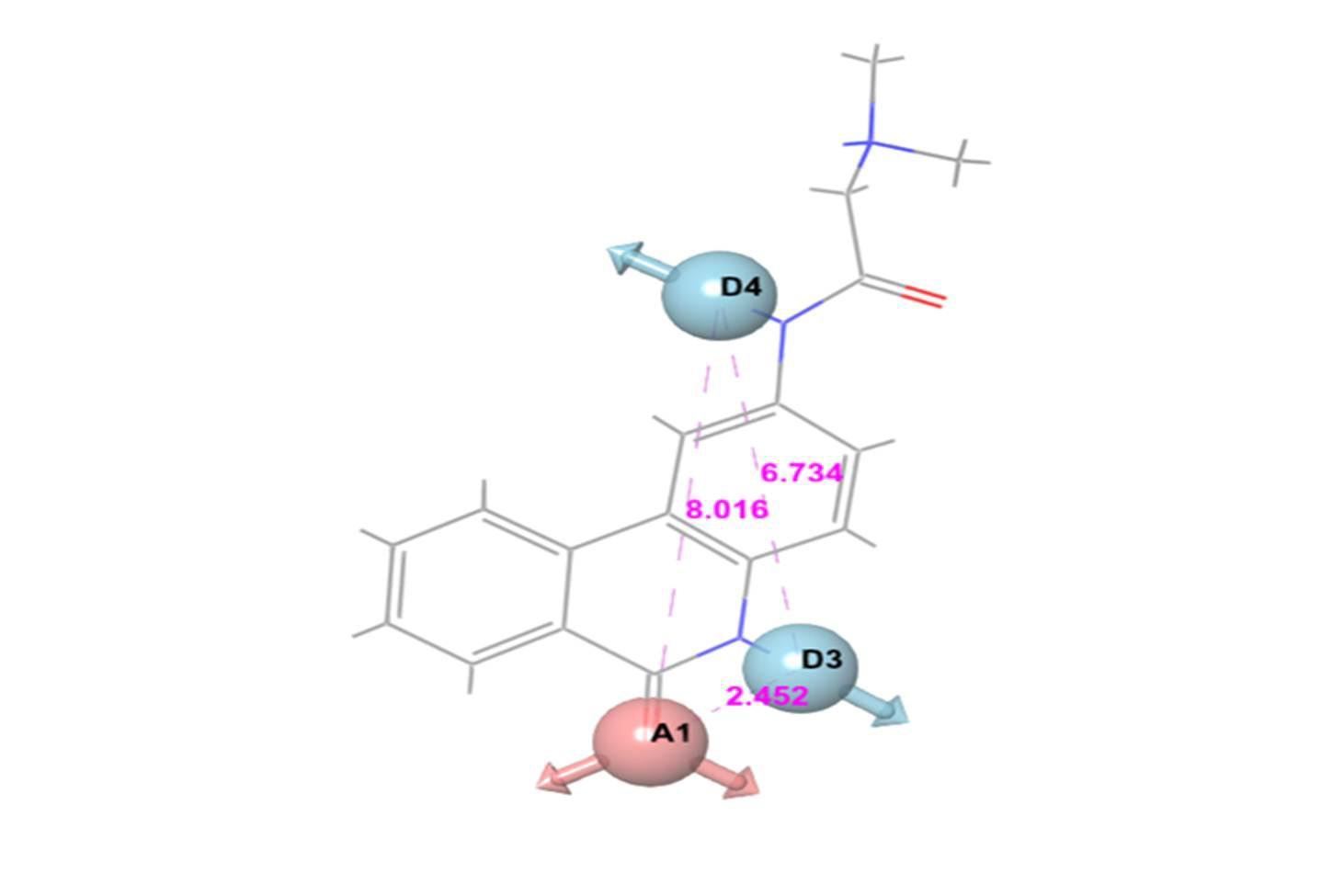
Figure 3: Three-point pharmacophore hypothesis of known Nucleocapsid (N) protein inhibitor with distance.
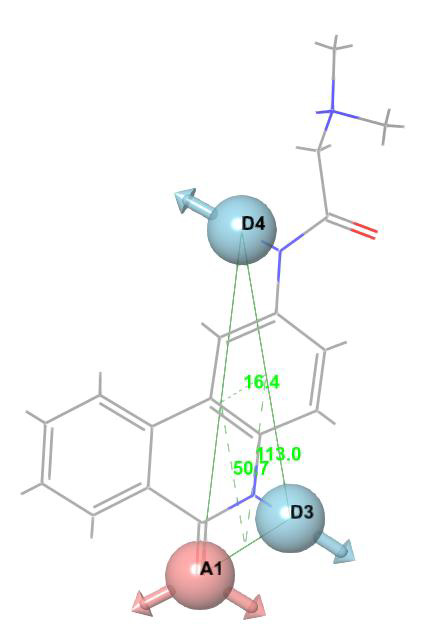
Figure 4: Three-point pharmacophore hypothesis of known Nucleocapsid (N) protein inhibitor with angles.
Prediction of ADME Properties
QikProp is a popular, fast, inexpensive, computational package that makes it easier to infer the pharmacokinetic properties of candidate drugs based on their molecular structures. The program predicts both physical significant descriptors and pharmaceutical pertinent traits. All the chemical entities were neutralized using QikProp application [21,22]. The default settings in the application were employed to determine the ADME properties of candidate molecules with desired pharmacokinetic properties.
Molecular Dynamics Simulation
Desmond simulation package was used to study the stability of docked SARS-CoV-2 Nucleocapsid (N) protein-ligand complex at a timescale of 100 ns [23]. Orthorhombic box was used to create a 10Å buffer region between the protein atoms and the box sides and the system was neutralized with Na and Cl ions. The OPLS3 force filed was used for energy minimization. The temperature was maintained constant at 300K, and a 2.0 fs values were obtained in the integration step [24-30]. Finally, molecular dynamics simulation was carried out at 100ns time scale for all four SARSCOV-2 Nucleocapsid (N) protein-hit compound complexes. The RMSD, RMSF and protein-ligand interactions were analyzed through simulation event analysis panel.
Results and Discussion
To evaluate the screening results, the reference ligand was downloaded from PubChem and docked it into the active site of Nucleocapsid (N) protein. During the docking interactions, it was observed that the amino acid residues GLN 189, THR 26 exhibited hydrogen bond interactions while LEU 27, CYS 145, MET 165 and MET 49 residues showed hydrophophic interaction with the reference compound PJ34. The Glide score and Glide energy of Nucleocapsid (N) protein-Reference ligand was -5.33 kcal/mol, -47.32kcal/mol respectively. The docking analysis initially resulted in screening of 500 compounds with HTVS. These compounds were further docked with the binding pocket of SARS-CoV-2 Nucleocapsid (N) protein in the SP mode. This led to identification of four potential ligands based on the Glide score. The four potential drug-like molecules had desirable pharmacokinetic properties and their structural representation was given below (Figures 5a-5d) (Table 1).
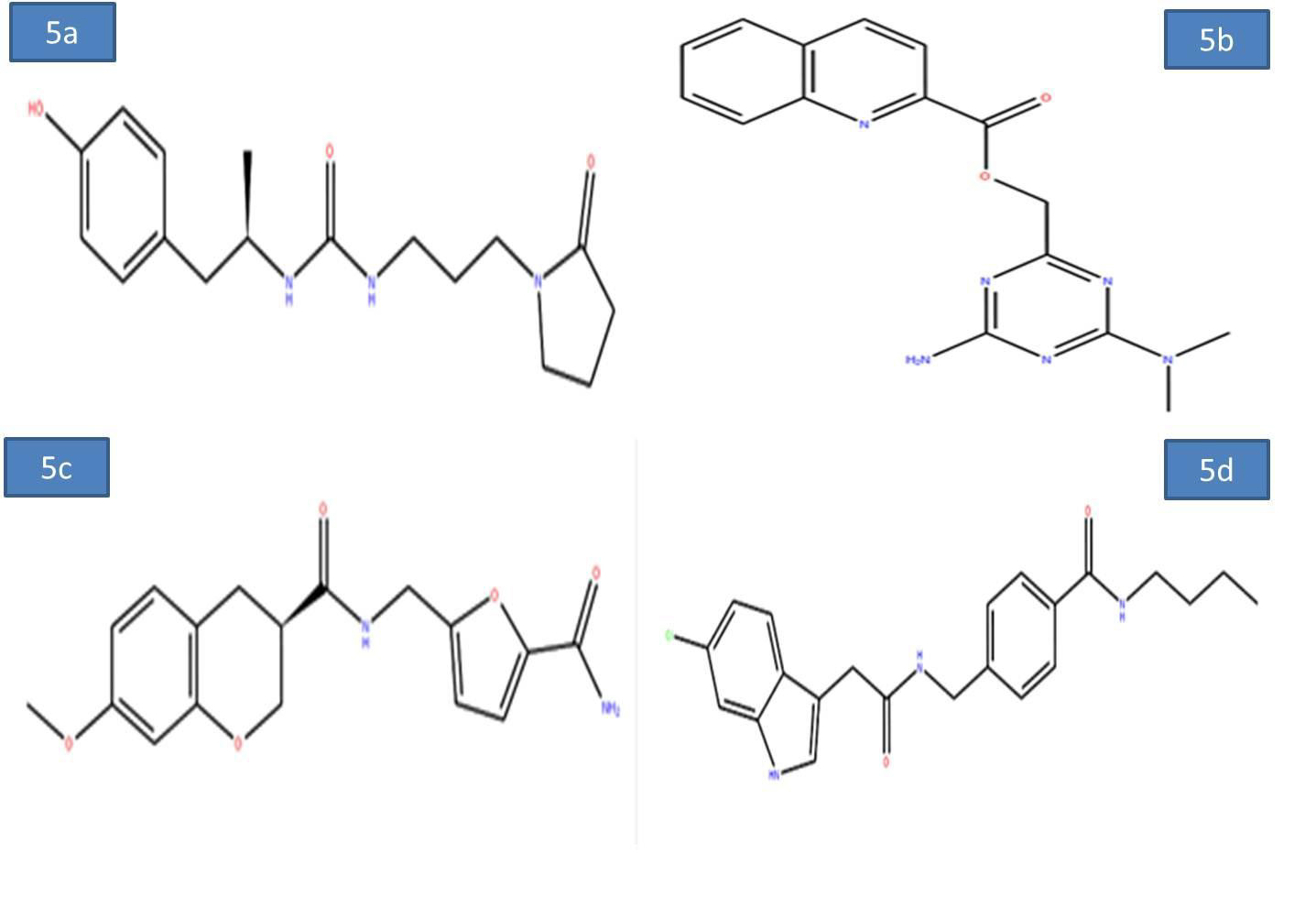
Figure 5a-d: 2D diagram of virtual screening hits a) ZINC55083699; b) ZINC12487932; c) ZINC81013764; d) ZINC68104820.
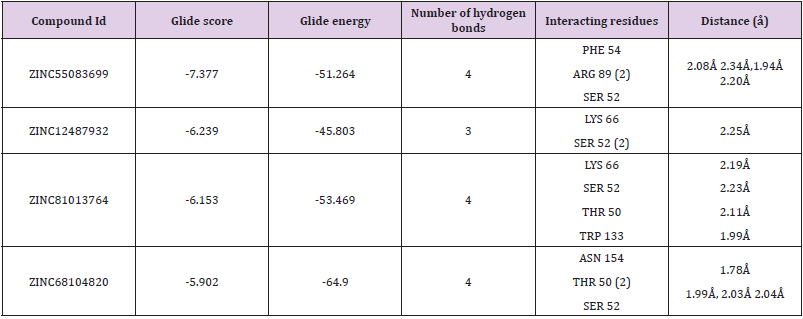
Table 1: Docking results of the four molecules.
Note: The IDs are of the ZINC database; Glide score (Kcal/mol); Glide energy (Kcal/mol); No of hydrogen bond interaction; Interacting residues; Distance between the protein and ligand (Å).
Docking Study
a. Binding Mode of ZINC55083699: The binding mode of ZINC55083699 indicates four hydrogen bond interactions within the active site of the SARS-CoV-2 Nucleocapsid (N) protein. The key amino acid residues of Nucleocapsid (N) protein involved in generating hydrogen bond network with ZINC55083699 compound includes PHE 54 (2.08Å), ARG 89 (2.34Å, 1.94Å), and SER 52 (2.20Å). The Glide score and Glide energy of the docked complex were -7.377kcal/mol and -51.264 kcal/mol respectively. Similarly, amino acids in the binding pocket such as ALA 157, VAL 159, ALA 91, TYR 112, PRO 169, and PRO 118 accounted for hydrophobic interactions. The crucial residues ARG 150, ARG 89 and LYS 66 were involved in the formation of pi-pi stacking and pi-cation interactions and all these interactions were responsible for increase in the binding stability of Nucleocapsid (N) protein- ZINC55083699 docked complex.
b. Binding Mode of ZINC12487932: The binding mode of ZINC12487932 in the active site of Nucleocapsid (N) protein indicates generation of hydrogen bonds with LYS 66 (2.25Å) and SER 52 (2.10Å, 2.13Å). The Glide score and Glide energy of the docked complex was found to be -6.239 kcal/mol and -45.803 kcal/ mol respectively. Similarly, PRO 68, ALA 51, VAL 159, TYR 110 and PRO 118 amino acids located in the binding pocket of Nucleocapsid (N) protein accounted for hydrophobic interactions.
c. Binding Mode of ZINC81013764: ZINC81013764 on binding in the active site of Nucleocapsid (N) protein exhibits three hydrogen bond interactions. The key amino acid residues involved in hydrogen bond includes LYS 66 (2.19Å), SER 52 (2.23Å), THR 50 (2.11Å) and TRP 133 (1.99Å) were involved in generating hydrogen bond network. The Glide scores and Glide energy of the docked complex were -6.153 kcal/mol and –53.469 kcal/mol respectively. The amino acids residues present at the binding pocket PRO 68, ALA 51, VAL 159, TYR 110, ILE 132 and TYR 112 were responsible for hydrophobic interactions in the docked Protein-ligand complex.
d. Binding Mode of ZINC68104820: The binding interaction of ZINC68104820 at the active site of Nucleocapsid (N) protein indicates generation of four hydrogen bonds due to the participation of Nucleocapsid (N) protein key amino acid residues ASN 154 (1.78Å), THR 50 (1.99Å, 2.03Å) and SER 52 (2.04Å). The Glide scores and Glide energy of the docked complex were -5.902 kcal/mol and -64.900 kcal/mol respectively. The hydrophobic interactions in the complex were due to ALA 51, PHE 54, ALA 56, TYR 112 and ALA 157 amino acid residues present at the binding pocket. Furthermore, interestingly only one pi-cation, pi-pi stacking interactions were observed with LYS 66 and ARG 150 amino acid residues (Figure 6).
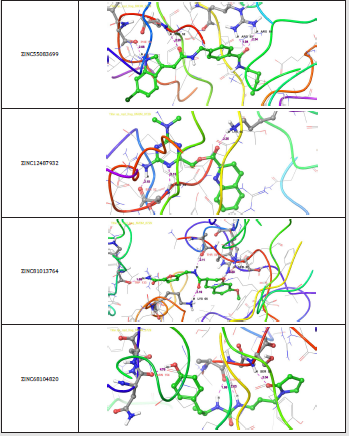
Figure 6: 3-D interaction representation of the top four hit compounds a) ZINC55083699 b) ZINC12487932 c) ZINC81013764 d) ZINC68104820 in the active site of the Nucleocapsid (N) protein.
Prediction of ADME Properties
ADME describes the destiny of drug like compounds within living organisms especially in the human system. The pharmacokinetics traits of four ligands were tested using Qikprop module. All the four molecules satisfied drug-like traits based on Rule of Five. Their molecular mass was smaller than 500kDa, with H-bond donor below 5, H-bond acceptor below 10 and LogP value less than 5 and follows Lipinski rule for drug-like molecules. Concurrently, these four molecules were further tested for their drug-like behaviour through investigation of PK factors mandatory for absorption, dispersal, biotransformation, elimination and toxicity. The aqueous solubility (QPLogS) vital for appraisal of absorption and distribution of drug molecule inside the body ranged for the four molecules ranged between -6.258 to -2.823 respectively. The drug metabolism and penetration of biological membrane were assessed using cell permeability method (QPPcaco2) shows a range from~ 122 to ~652. The predicted value of QPPMDCK ranged from to 162 to 1126. The predicted values of binding to human serum albumin (QPlogksha) were within the acceptable ranges from -0.058 to 0.358. The predicted Brain/Blood barriers were under tolerable range from ~ -1.536 to -0.981, the predicted hydrophobic SASA were under acceptable range of 372.631 to 186.067. The predicted rotatable bonds were well with acceptable range 5.000 to 8.000. The % range of human oral absorption in GI was from 73 to 100. The predicted ADME properties results were displayed in the (Table 2).
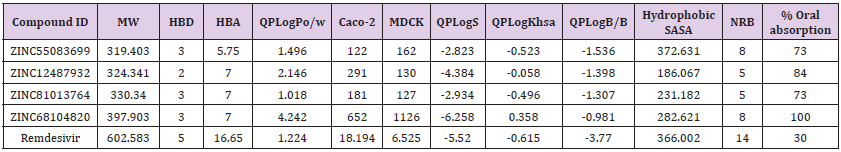
Table 2: Drug-like properties of the new chemical entities determined by Qikprop.
Note: Molecular weight (under 500 Da); Hydrogen bond donors (lee then 5); Hydrogen bond acceptors (not more then 10); QP log P for octanol/water ( -2.0 / 6.5); Apparent Caco-2 Permeability (nm/sec) (<25 poor, >500 great); Apparent MDCK Permeability (nm/ sec) (<25 poor, >500 great); QP log S for aqueous solubility (-6.5 / 0.5); QP log K hsa Serum Protein Binding (acceptable range; -1.5 to 1.5); QP log BB for brain/blood ( -3.0 / 1.2); Solute Hydrophobic SASA (acceptable range; 0.0 to 750.0); Solute No. of Rotatable Bonds (acceptable range; 0.0 to 15.0); % Human Oral Absorption in GI (+-20%) (<25% is poor).
Molecular Dynamics (MD) Simulation
a) MD Analysis of Sars-Cov-2 Nucleocapsid (N) Protein - Zinc55083699 Complex: The binding stability of SARS-CoV-2 Nucleocapsid (N) protein–ZINC55083699 complex was examined using molecular dynamics simulations. The RMSD of protein-ligand complex was reported in figure 7a. At 15ns, a slight deviation was observed in c-alpha with an RMSD of 6.38Å and became stable till entire simulation with an observed RMSD value of 5.97Å. The root-mean-square fluctuations (RMSF) of the SARS-CoV-2 Nucleocapsid (N) protein were represented in figure 7b. The Cα, backbone and side chains displayed high RMSF values at GLY 98, ASP 99 with observed RMSF values as 10.86Å, 10.14Å and 10.50Å, respectively. The key residues THR 50, ASN 49 and ASP 99 in Cα backbone and side chains of SARS-CoV-2 Nucleocapsid (N) protein exhibited lower degree of fluctuations with RMSF values 2.54Å, 4.68Å and 9.98Å respectively. The bar diagram of 7c represents SARS-CoV-2 Nucleocapsid (N) protein-ZINC55083699 interactions. Hydrogen bond interactions were observed with ASN 70, TYR 124 amino acid residues and the occupancy was noted 11.8%, and 33.7% respectively. The overall Nucleocapsid (N) protein-ligand interactions observed during the simulation presented in (Figure 7d). This indicates that key amino acid residues were involved in the formation of hydrogen bond (ASN 78, ALA 156 and ASN 76 for 13%, 13% and 20%,) and water bridge contact (TYR 124 for 15%) with Nucleocapsid (N) protein leading to increased binding stability of the Nucleocapsid (N) protein-ligand complex during the simulation time (Figure 7).
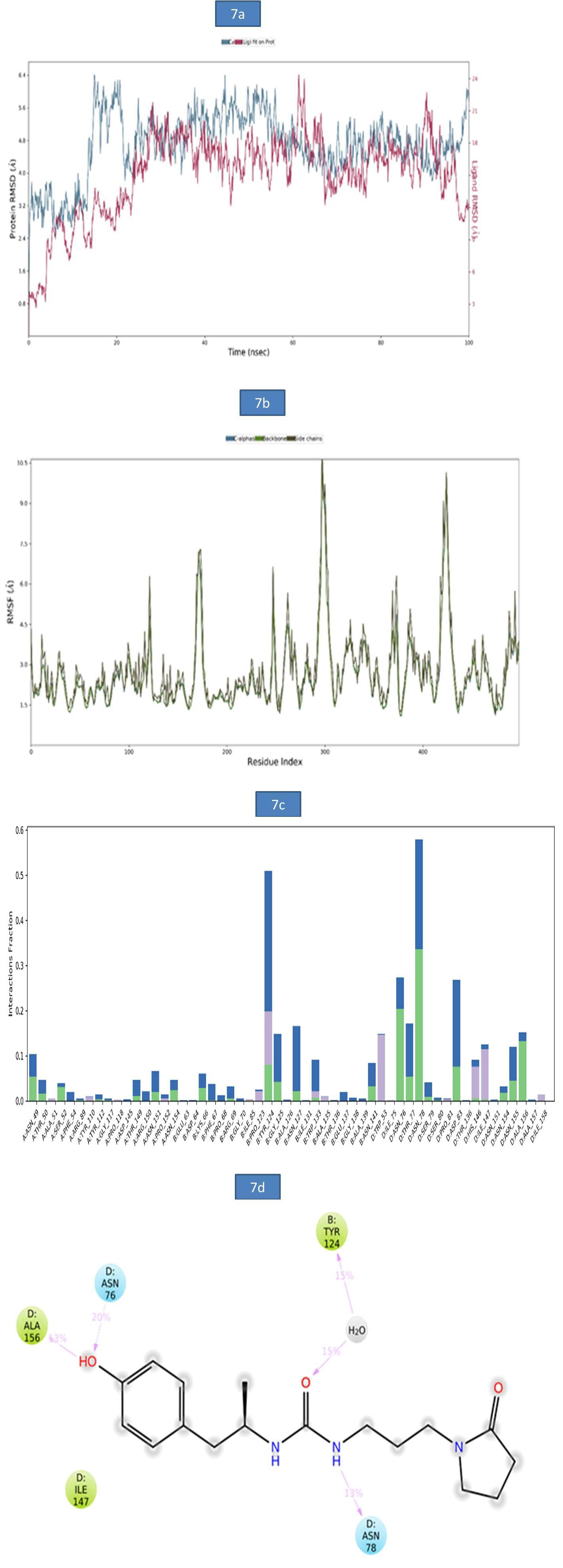
Figure 7: a). The RMSD plot of SARSCOV-2 Nucleocapsid (N) protein-ZINC55083699 complex during 100-ns simulations. b). The RMSF plot of SARSCOV-2 Nucleocapsid (N) protein during the 100ns simulations. c) The RMSF map of SARSCOV-2 Nucleocapsid (N) protein - ZINC55083699 complex over the 100ns simulations. d). The bar diagrams of SARSCOV-2 Nucleocapsid (N) protein - ZINC55083699 contacts during 100ns simulations.
b) MD analysis of SARS-CoV- Nucleocapsid (N) protein – ZINC12487932 complex: The RMSD of protein-ligand complex was reported in Figure 8a. There was no deviation observed in the protein RMSD but, a slight deviation was observed in ligand RMSD at 61.40 ns (5.04Å), 75ns (6.86Å) and 99.90ns (6.59Å). The rootmean- square fluctuations (RMSF) of the SARSCOV-2 Nucleocapsid (N) protein were represented in figure 8b. The Cα, backbone and side chain displayed high RMSF values in the position of GLY 98 (10.49Å), GLY 98 (10.18Å) and ASP 99 (10.17Å) respectively. The bar diagram of 8c represents SARSCOV-2 Nucleocapsid (N) protein- ZINC12487932 interactions. The hydrogen bond interactions were observed with SER 52, TYR 112 and the occupancy was noted as 56% and 61.4% respectively. The key amino acid residues SER 52, PHE 54 and TYR 112 were involved in the formation of hydrogen bond to the extent of 46%, 37% and 31% respectively. A water bridge contact with LYS 66 of target protein was observed as 42% and also ligand formed only one pi-pi stacking interaction with TYR 110. These molecular interactions at the binding site region of the SARSCOV-2 Nucleocapsid (N) protein lead to the improved binding stability of the Nucleocapsid (N) protein-ZINC12487932 complex during simulation time. (Figure 8).
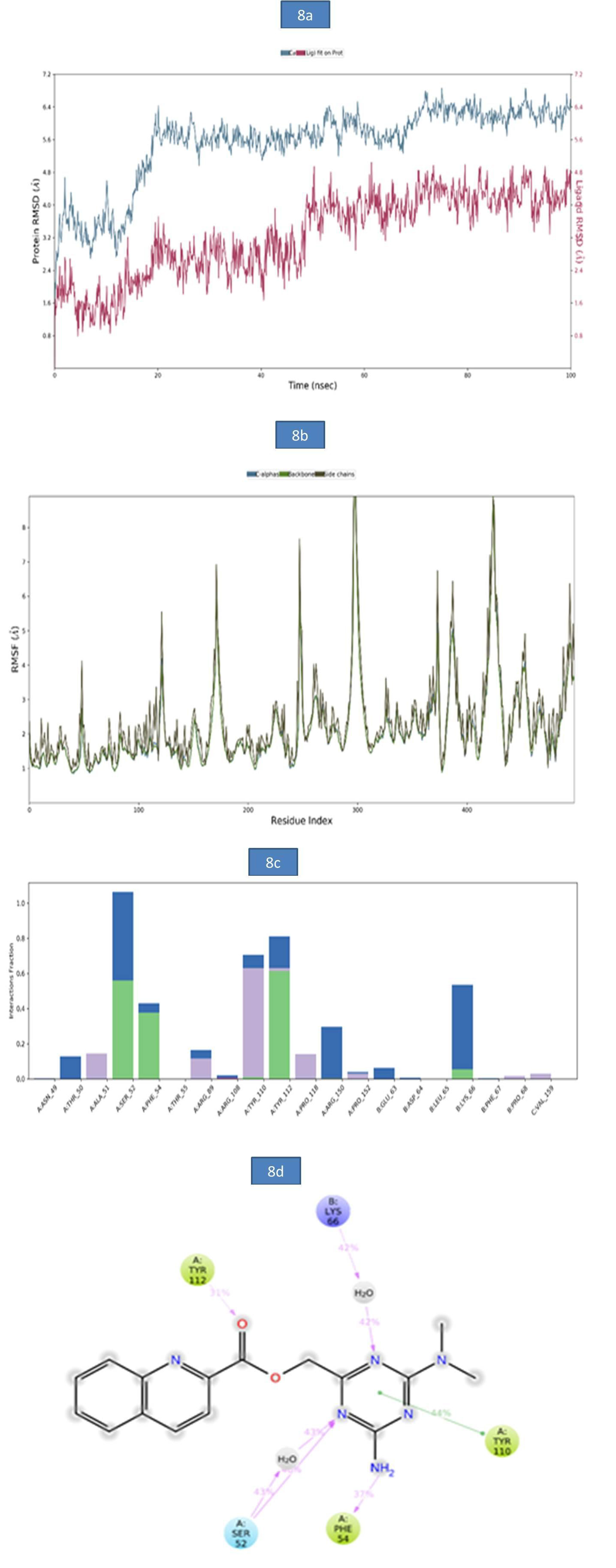
Figure 8: a). The RMSD plot of SARSCOV-2 Nucleocapsid (N) protein-ZINC12487932 complex during 100-26 ns simulations.
b). The RMSF plot of SARSCOV-2 Nucleocapsid (N) protein during the 100ns simulations. c) The RMSF map of SARSCOV-2 Nucleocapsid (N) protein - ZINC12487932 complex over the 100ns simulations. d). The bar diagrams of SARSCOV-2 Nucleocapsid (N) protein - ZINC12487932 contacts during 100ns simulations.
c) MD Analysis of Sars-Cov-2 Nucleocapsid (N) Protein – Zinc81013764 Complex: The RMSD of Nucleocapsid (N) protein -ligand complex was reported in figure 9a. A slight RMSD deviation was observed in c-alpha at 37.20ns followed by a more stable RMSD value of 5.79Å till the end of entire simulation. The root-meansquare fluctuations (RMSF) of the SARSCOV-2 Nucleocapsid (N) protein was represented in (Figure 9b). The Cα, backbone and side chain displayed high RMSF values in the amino acid residues of GLY 100, ASP 99, GLY 98 with RMSF values 10.54Å, 10.20Å and 11.54Å, respectively. The key residues GLY 98, THR 50 and MET 102 in the Cα atoms, backbone and side chain SARSCOV-2 Nucleocapsid (N) protein exhibited lower degree of fluctuations with RMSF values 8.58Å, 2.27Å and 6.20Å respectively. The bar diagram of 9c were represent the SARSCOV-2 Nucleocapsid (N) protein-ZINC81013764 interactions. A hydrogen bond interaction was observed with SER 32, TYR 110 and the occupancy was noted 75.9% and 76.6% respectively. In addition, figure 9d indicates key amino acid residues SER 52 and ASN 154 involved in the formation of hydrogen bond (52% and 54%) and water bridge contact ARG 89(82%), TYR 112 (80%) with Nucleocapsid (N) protein and one pi-cation interaction with LYS (66 44%). The molecular interactions contributed to the enhanced binding stability of the protein-ligand complex during the simulation time (Figure 9).
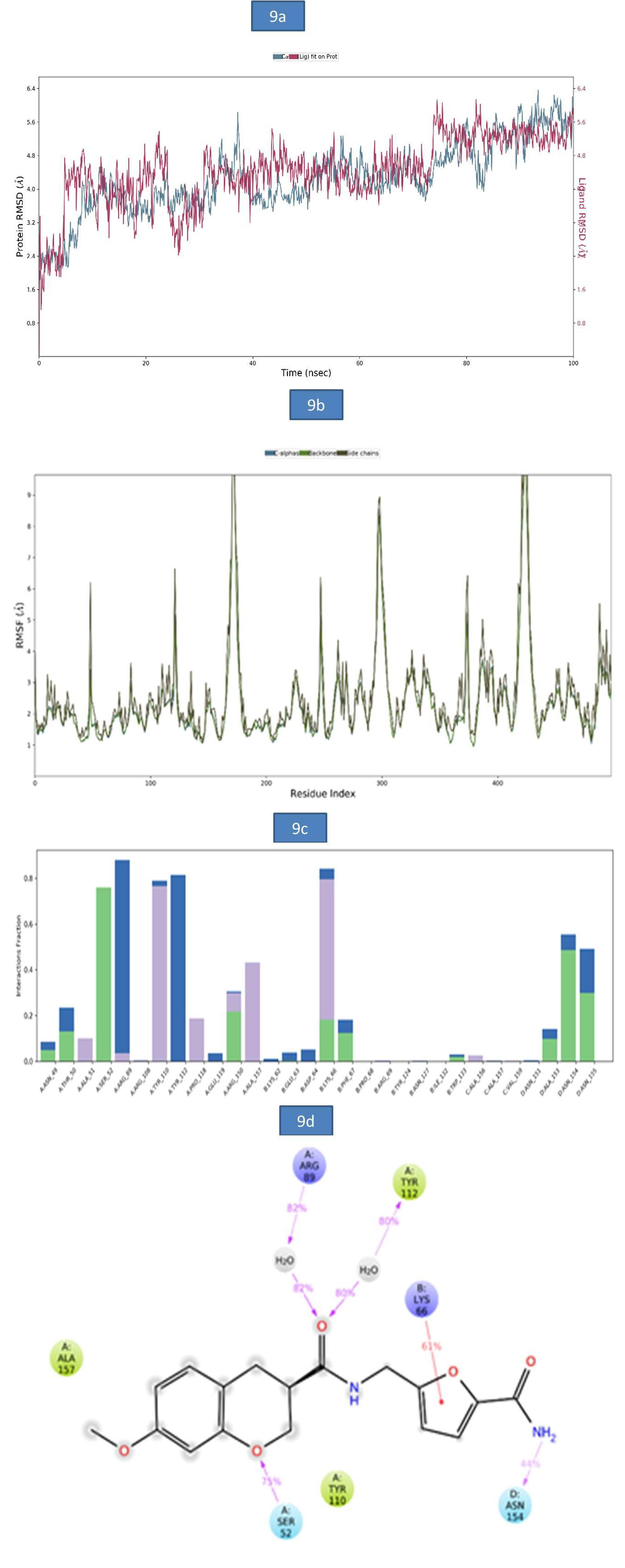
Figure 9: a). The RMSD plot of SARSCOV-2 Nucleocapsid (N) protein - ZINC81013764 complex during 100-ns simulations.
b). The RMSF plot of SARSCOV-2 Nucleocapsid (N) protein during the 100ns simulations. c) The RMSF map of SARSCOV-2 Nucleocapsid (N) protein-ZINC81013764 complex over the 100ns simulations. d). The bar diagrams of SARSCOV-2 Nucleocapsid (N) protein - ZINC81013764 contacts during 100ns simulations.
d) MD Analysis of Sars-Cov-2 Nucleocapsid (N) Protein- Zinc68104820 Complex: The RMSD of protein-ligand complex was given in Figure 10a. The RMSD of the c-alpha was more stable during the entire simulation period and the RMSD observed was 7.73Å. Ligand RMSD exhibited slight deviation at 25ns- 31.20ns and the RMSD values observed were 8.14 to 8.28Å respectively and was stable during the remaining simulation period was with RMSD reaching 4.08Å. The root-mean-square fluctuations (RMSF) of the SARSCOV-2 Nucleocapsid (N) protein–ZINC68104820 were presented in figure 10b. The Cα, backbone and side chain displayed high RMSF values in the position of GLY 98 (8.65Å). Hydrogen bond interactions were observed with ARG 150 and the occupancy 74.9% occupancy value. In addition, the key amino acid residue ARG 150 was involved in the formation of pi-pi stacking (38% and 42%) and water bridge contact (43%) with target protein. These molecular interactions contributed to the stability of the Nucleocapsid (N) protein-Ligand complex during the simulation period (Figure 10).
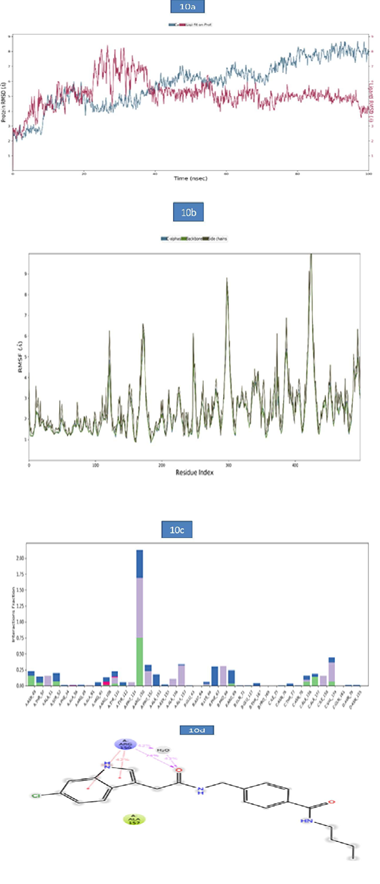
Figure 10:
a. The RMSD plot of SARSCOV-2 Nucleocapsid (N) protein - ZINC68104820 complex during 100-ns simulations.
b. The RMSF plot of SARSCOV-2 Nucleocapsid (N) protein during the 100ns simulations.
c. The RMSF map of SARSCOV-2 Nucleocapsid (N) protein - ZINC68104820 complex over the 100ns simulations.
d. The bar diagrams of SARSCOV-2 Nucleocapsid (N) protein - ZINC68104820 contacts during 100ns simulations.
Conclusion
Nucleocapsid (N) protein is an essential structural protein mainly responsible for the interaction, transcription, and invasion of the SARS-CoV-2 viral genome and is considered as an attractive drug target for the treatment of COVID-19. In the current study, four potential Nucleocapsid (N) protein inhibitors viz., ZINC55083699, ZINC12487932, ZINC81013764 and ZINC68104820 were identified through E-pharmacophore from the zinc database. All the Pharmacokinetic properties of these 4 compounds met the required criteria. Molecular Dynamics simulation for 100 ns revealed all the 4 compounds to be stable in the solvent environment with respect to distance and fluctuation dynamics. These 4 compounds on further studies could be used as novel leads for the inhibition of Nucleocapsid (N) protein.
Author Contributions
Conceived and designed the experiments: AM, VRT, KE, AJ, PV. Performed the experiments: AM. Analyzed the data: AM, KE, VRT, PV. Contributed software facility: AM. Wrote the paper: AM, H.-F.J., KE. Assisted in writing the paper and referencing: H.-F. J, WJ, VRT.
Conflict of Interest
The authors declare that they have no competing interests.
Compliance with Ethical Standards
Not applicable. The ethical standards have been met.
Funding
This compilation is a research article written by its author and required no substantial funding to stated.
Data Availability
From corresponding authors upon request.
References
- Li X, Geng M, Peng Y, Meng L, Lu S (2020) Molecular immune pathogenesis and diagnosis of COVID-19. J Pharm Anal 10(2): 102‐108.
- https://www.who.int/emergencies/diseases/novel-coronavirus-2019?
- Hoffmann M, Kleine Weber H, Schroeder S, Nadine Krüger, Tanja Herrler, et al. (2020) SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 181(2): 271-280.
- Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, et al. (2020) Structure, function, and antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 181(2): 281-292.
- Fehr AR, Perlman S (2015) Coronaviruses: An Overview of Their Replication and Pathogenesis. In: Maier H, Bickerton E, Britton P (Eds.)., Methods in Molecular Biology 1282. Humana Press, New York, NY.
- Wan S, Xiang Y, Fang W, Yu Zheng, Boqun Li, et al. (2020) Clinical Features and Treatment of COVID-19 Patients in Northeast Chongqing. J Med Virol, p. 1-10.
- XuY (2020) Unveiling the Origin and Transmission of 2019-nCoV. Trends Microbiol 28(4): 239-240.
- Wu A, Peng Y, Huang B, XiaoDing, XianyueWang, et al. (2020) Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. Cell Host Microbe 27(3): 325-328.
- Simran Preet Kaur, Vandana Gupta (2020) COVID-19 Vaccine: A comprehensive status report. Virus Research 288: 198114.
- Malik YS, Sircar S, Bhat S, Khan Sharun, Kuldeep Dhama, et al. (2020) Emerging novel coronavirus (2019-nCoV)-current scenario, evolutionary perspective based on genome analysis and recent developments. Vet Q 40(1): 68-76.
- Ding Q, Lu P, Fan Y, Xia Y, Liu M (2020) clinical characteristics of pneumonia patients coinfected with 2019 novel coronavirus and influenza virus in Wuhan, China. J Med Virol 92(9): 1-7.
- Umesh Kundu D, Selvaraj C, Singh SK, Dubey VK (2020) Identification of new anti-nCoV drug chemical compounds from Indian spices exploiting SARS-CoV-2 main protease as target. J Biomol Struct Dyn, p. 1-9.
- Bhardwaj VK, Singh R, Sharma J, Rajendran V, Purohit R, et al. (2020) Identification of bioactive molecules from Tea plant as SARS-CoV-2 main protease inhibitors. J Biomol Struct Dyn 39(10): 1-13.
- Mahanta S, Chowdhury P, Gogoi N, Nabajyoti Goswami, Debajit Borah, et al. (2020) Potential anti-viral activity of approved repurposed drug against main protease of SARS-CoV-2: an in silico-based approach. J Biomol Struct Dyn 39(10): 1-15.
- Khan MT, Ali A, Wang Q, Muhammad Irfan, Abbas Khan, et al. (2020) Marine natural compounds as potents inhibitors against the main protease of SARS-CoV-2-A molecular dynamic study. J Biomol Struct Dyn, p. 1-14.
- Kang S, Yang M, Hong Z, Liping Zhang, Zhaoxia Huang, et al. (2020) Crystal structure of SARS-CoV-2 nucleocapsid protein RNA binding domain reveals potential unique drug targeting sites. Acta Pharm Sin B 10(7):1228-1238.
- Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W (2013) Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des 27(3): 221‐234.
- (2009) Protein preparation Wizard Maestro. New York: Schrodinger LLC.
- (2009) Glide, Version 5.5 (2009) Schrodinger, LLC, New York, NY.
- Pérez Regidor L, Zarioh M, Ortega L, Martín-Santamaría S (2016) Virtual Screening Approaches towards the Discovery of Toll-Like Receptor Modulators. Int J Mol Sci 17(9): 1508.
- Dik LM, Daniel SC, Chung HL (2011) Molecular docking for virtual screening of natural product databases. Chem Sci 2(9): 1656-1665.
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv drug deli Rev 23(1-3): 3-25.
- (2010) QikProp, Version 2.3, LLC, New York, NY.
- Duffy EM, Jorgensen WL (2000) Prediction of properties from simulations: free energies of salvation in Hexa decane, octanol, and water. J Am Chem 122: 2878-2888.
- Ganesh Kumar Veeramachaneni, K Kranthi Raj, Leela Madhuri Chalasani, Jayakumar Singh Bondili, Venkateswara Rao Talluri (2015) High-throughput virtual screening with e-pharmacophore and molecular simulations study in the designing of pancreatic lipase inhibitors. Drug Des Devel Ther 9: 4397-4412.
- Lin SY, Liu CL, Chang YM, Zhao J, Perlman S, et al. (2014) Structural basis for the identification of the N-terminal domain of coronavirus nucleocapsid protein as an antiviral target. J Med Chem 57(6): 2247-2257.
- Sunghwan K, Paul AT, Evan E B, Jie Chen, Gang Fu, et al. (2016) PubChem Substance and Compound databases. Nucleic Acids Res 44: D1202-D1213.
- Ligprep, Version 2.3, Schrodinger, LLC, New York, NY.
- Ramin Ekhteiari Salmas, Ayhan Unlu, Muhammet Bektaş, Mine Yurtsever, Mert Mestanoglu et al. (2017) Virtual Screening of Small Molecules Databases for Discovery of Novel PARP-1 Inhibitors: Combination of in silico and in vitro J Biomol Struct Dyn 35(9): 1899-1915.
- Subramaniyan Vijayakumar, Palani Manogar, Srinivasan Prabhu, Ram Avadhar Sanjeevkumar Singh (2018) Novel ligand-based docking; molecular dynamic simulations; and absorption, distribution, metabolism, and excretion approach to analyzing potential acetylcholinesterase inhibitors for Alzheimer's disease. J Pharm Anal 8(6): 413-420.
