 Research Article
Research ArticleAbstract
A new and modern method for investigation of covid-19 has been reported. In this study, simulation between coronavirus with the synthesized compounds as new drugs such as N-(glycine)-para styrene sulfonamide (GSS) and N-(alanine) - Parastyrene sulfonamide (ASS) was performed and compared with sulphadiazine (SDA), sulfacetamide (SAC), and sulfathiazole (STZ) as common drugs. Molecular docking has recently been used as a tool to gain insight into ligand–receptor interaction and display molecules for the binding affinities against a special receptor. Molecular docking calculations were performed on Auto Dock-Vina software. The 3D crystal structure of employed SARS-CoV spike glycoprotein (7ACD) as Covid-19 and receptor were obtained from Protein Data Bank. The empirical free energy and the Lamarckian Genetic Algorithm was applied for molecular docking 7ACDwith GSS and ASS as anti-infection agents was used for molecular docking simulation. In addition, these interactions were compared with sulphadiazine, sulfacetamide, and sulfathiazole modeling. That is shown biological properties of these new sulfonamides similar to sulphadiazine, sulfacetamide, and sulfathiazole.
Keywords: N-(glycine)-Para Styrene Sulfonamide; N-(alanine) - Para- Styrene Sulfonamide; 7ACD; Coronavirus; Sulphadiazine; Sulfacetamide; Sulfathiazole
Introduction
COVID-19 has been identified as a worldwide epidemic virus that causes an acute respiratory syndrome [1]. The virus (SARSCoV- 2) was first identified in December 2019 in Wuhan, China. The outbreak of coronavirus in China and other countries has alarmed the international community. The World Health Organization announced the prevalence and urgency of public health on January 20, 2020. Approximately, more than 2 million deaths have been attributed to COVID-19, making it one of the deadliest pandemics in history. In the first, no vaccine or drug was available to destroy the virus [2]. Since the first case of a novel coronavirus (COVID-19), infection pneumonia was discovered in Wuhan, China, a series of confirmed cases of the COVID-19 were found in Beijing [3]. Due to the lack of immunity of the human body to this virus, which causes cell death and damage caused by it, the use of drugs to eradicate this illness is of special value [4]. Several drugs such as chloroquine, arbidol, remdesivir, and favipiravir used for the treatment of coronavirus disease [5] in the other ward, Glycan shield and structure prediction of spike glycoprotein and its interaction with human CD26 in the simulation method, was reported [6] that predict the structure of COVID- 19 and significant its interaction with HIV-1 Tat protein derived N-terminal nonapeptide Trp2-Tat (1-9) bound to the active site of Dipeptidyl peptidase IV (CD26) [6]. In following these research, therapeutic strategies were recommended including: anti-viral [7-10] anti-inflammatory [11] drugs, antimalaria [12], and antirheumatic drugs [13]. This research made us eager that recommend the sulfonamide group as a useful and new drug and simulating them with 7ACD (COVID-19), as Spike glycoprotein. The aim of this article is to suggest the effectiveness of these drugs against COVID-19. We discuss the approaches for developing therapeutic to cope with this viral outbreak. Molecular docking was completed to identify the interaction of ASS and GSS with dimeric MERS-CoV spike glycoprotein (7ACD). Then, we have been found that ASS and GSS possess anti- infective properties.
Experimental
Preparation the N-(glycine) - Para- Styrene Sulfonamide (GSS)
The synthetic method for the preparation of the GSS with chemical formula (C10H11NO4S) is as follows: In a 100 mL round bottomed flask, equipped with magnetic stirrer, 4.86 g para vinyl styrene sulfonyl chloride (24 mmol) in 50 mL CHCl3 as a solvent and 1.8 g glycine (24 mmol) were added. Then, 24 mL NaOH 1M was slowly added. Experimental details of GSS are described in Ref [14].
Preparation of N-(alanine) - Para- Styrene Sulfonamide (ASS)
The synthetic method for the preparation of the ASS with chemical formula (C11H13NO4S) is as follows: in a 100 mL roundbottomed flask, equipped with magnetic stirrer, 4.86 g p-styrene sulfonyl chloride (24 mmol) in 50 mL CHCl3 as a solvent and 2.14 g (S)-(+)-alanine (24 mmol) were placed. Then, 2.4 mL NaOH 1M was slowly added. Experimental details of ASS are described in Ref [15].
Computational Details
In the first step, the optimization of the ASS and GSS was performed using Gaussian 09 software at the Becke3- Lee-Yangparr (B3LYP) method level with a 6-311+G (d, p) basis set [16]. Secondly, the output of the geometry optimization for these ligands was applied for the docking process. In this work, the 3D Crystal structure of the dimeric MERS-CoV receptor-binding domain (PDB ID: 7ACD) was taken from the Brookhaven Protein Data Bank. Molecular docking calculations were performed on Auto Dock- Vina software [17] Lamarckian Genetic Algorithm (LGA) available in Auto dock was employed for docking, as the most popular algorithm [18,19]. The graphical representation of ligand-receptor interaction was obtained using ligplot software [20].
Results and Discussion
Molecular Structural
The optimized geometry (opt-freq) using B3LYP / 6-311+G (d, p) of N-(glycine) - Para- styrene sulfonamide (GSS), and N-(alanine) - Para- styrene sulfonamide (ASS) were shown in Figure 1.
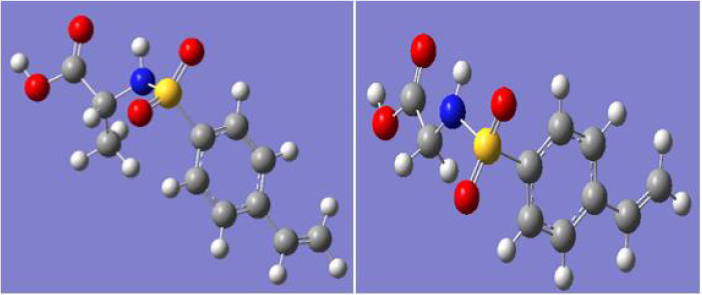
Figure 1: The geometry of ASS in the left, and GSS in the right.
Molecular Electrostatic Potential
MEP (Molecular electrostatic potential) is a significant tool for predicting electrophilic and nucleophilic attacks for biological interactions [21,22]. The MEP of sulphadiazone (SDA), sulfacetamide (SAC), and sulfathiazole (STZ) were optimized geometry using B3LYP method 6-311G + (d, p). As can be observed in Figure 1. The different colors in this plot are indicated different values of the electrostatic potential. Red < orange < yellow< green < blue. The blue illustrates the strongest attraction. The positive area is located around CH groups. These areas have positive potential. The negative area is related to S=O groups. These areas having negative potential are over the electronegative atoms such as oxygen. The red color indicates the strongest repulsion. These regions of negative potential are associated with the lone pair of electronegative atoms.
The Disk Diffusion Method
A microbial suspension (1 ml) Staphylococcus aureus and Escherichia coli were applied separately over the surface of the agar plate, which was then incubated for 24 h at 37 °C in an autoclave. Inhibitory zone values (diameter of inhibition) from disk diffusion tests and growth inhibition ring for PSS are reported in ref [14,15]. GSS and ASS biological studies showed that both Staphylococcus aureus and Escherichia coli were susceptible to ASS and GSS. The results of biological research show that ASS and GSS can be used to design and synthesize new drugs. Following this research, by modeling these sulfonamide compounds with coronavirus, we introduce these compounds as suitable drugs for the treatment of this virus as anti-infective agents.
Molecular Docking Studies
We decided to perform molecular docking simulation of the GSS, SDZ, and ASS against the 3D crystal structure of dimeric MERSCoV spike glycoprotein was obtained from Protein Data Bank (PDB ID: 7ACD). Molecular docking is a significant investigation to understand the ligand- receptor interactions. The ligand was prepared for docking by B3LYP method 6-311+G (p,d)basis set. The active sites of the 7ACD were defined to include residues of the active site within the grid box size of 94A°×108A°×126A° for GSS, 76A°×122A°×126A° for ASS, 68A°×112A°×126A° for SDA, 76A°×92A°×126A° for SAC, and 75A°×91A°×126A° for STZ with a grid-point spacing of 1.00 Å were applied. Among the docked conformations, the best scored conformation predicted by Auto Dock scoring function were visualized for GSS-7ACD, ASS-7ACD, STZ–7ACD SDZ-7ACD, and SAC-7ACD interactions in Ligplot and Ligplus software. The resulting docking in which the ASS binds into the 7ACD creates two hydrogen bonds. These hydrogen bonds are between both of OCO2 of ASS with SER64 and TYR256 (2.92Ȧ, 2.94Ȧ) respectively. The GSS creates binds into the 7ACD creates three hydrogen bonds. These hydrogen bonds are between the OSO2 of GSS that one of oxygen with Asp560 (3.16 Ȧ) and other oxygen withThr559 (2.85 Ȧ, 3.04 Ȧ) and also NNH of GSS with Ser574 (2.98Ȧ) respectively. The SDA creates two hydrogen bonds. These hydrogen bonds are between Oso2 with Thr67 and NNH2 with Ser 64 (2.85 Ȧ, 3.26 Ȧ) respectively. The SAC creates four hydrogen bonds. Two hydrogen bonds are between Oso2 and NNH of SAC with Lys 198 (3.14 Ȧ, 2.96 Ȧ), the other two hydrogen bonds Oco of SAC with Arg183 (3.145 Ȧ, 3.18 Ȧ) respectively. The STZ creates two hydrogen bonds. These hydrogen bonds are between Oso2 and NNH with Tyr 256 (2.88 Ȧ, 3.23 Ȧ) respectively (Figure 2), viz. The binding free energy (ΔG° in kcal mol-1) -5.9 for GSS, -6.4 for ASS, -5.7 for SDA, -6.7 for SAC, and -6.8 for STZ are predicted for the best conformation of these compounds (Figure 3).
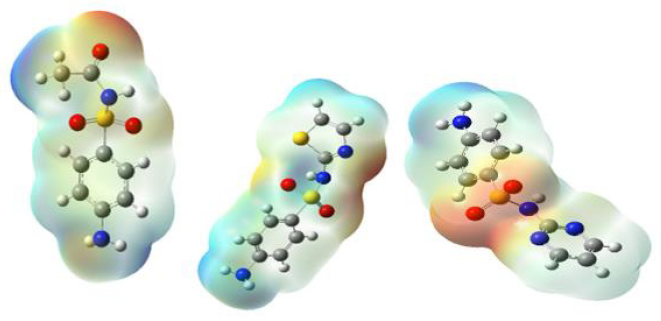
Figure 2: MEP plot SDA (in the left) and SAC in the middle STZ (in the right).
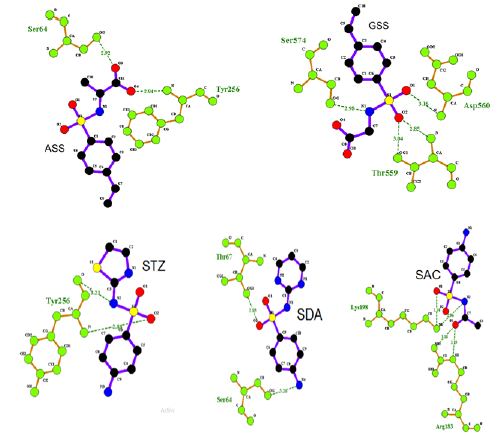
Figure 3: The interactions and hydrogen bonding across the binding interface of ASS–7ACD in the left top, GSS-7ACD in the right top, STZ–7ACD in the left bottom, SDA–7ACD in the middle bottom, SAC-7ACD in the right bottom and (H bonds are shown by green dotted lines).
Conclusion
We report the new method and modeling for the treatment of COVID – 19. This research confirms the anti- infective properties of ASS and GSS compounds that have similar to sulphadiazne, sulfacetamide, and sulfathiazole structures. These modeling to show, ASS, GSS, SDA, SAC, and STZ have interaction with COVID – 19. These biological investigation results suggest ASS and GSS can be used for the design and synthesis of the new based-drug materials in order to coronavirus treatment. In addition, SDA, SAC, and STZ also have these properties.
Acknowledgment
We would like to thank Lorestan University for its financial support.
References
- Peiris JS, M Guan, Y Yuen K (2004) Severe acute respiratory syndrome. Nat Med 10(12): S88-S97
- Renyi Wu, Lujing Wang, Hsiao-Chen Dina Kuo, Ahmad Shannar, Rebecca Peter, et al. (2020) An update on current therapeutic drugs treating COVID-19. Curr Pharmacol Rep 6(3): 56-70.
- Sijia Tian, Nan Hu, Jing Lou, Kun Chen, Xuqin Kang, et al. (2020) Characteristics of COVID-19 infection in Beijing. J Infect 80(4): 401- 406.
- Susanna Felsenstein, Jenny A Herbert, Paul S McNamara, Christian M Hedrich (2020) COVID-19: Immunology and treatment options. Clin Immunol: 108448.
- Dong L, Hu S, Gao J (2020) Discovering drugs to treat coronavirus disease 2019 (COVID-19). Drug discov Ther 14 (1): 58-60.
- Vankadari N, Wilce JA (2020) Emerging COVID-19 coronavirus: glycan shield and structure prediction of spike glycoprotein and its interaction with human CD26. Emerg Microbes & Infect 9(1): 601-604.
- Shafiee S, Davaran S (2020) A mini-review on the current COVID-19 treatments. Chem Rev Lett 3(1): 19-22.
- Jean S, S Hsueh PR (2020) Old and re-purposed drugs for the treatment of COVID-19. Expert Rev Anti-Infect Ther 18(9): 843-847.
- Pyrc K, Berkhout B, Van Der Hoek L (2007) Identification of new human coronaviruses. Expert Rev Anti-Infect Ther 5(2): 245-253.
- Yavuz S, Ünal S (2020) Antiviral treatment of COVID-19. Turk J Med Sci 50: 611-619.
- Zhang W, Zhao Y, Zhang F, Wang Q, Li T, et al. (2020) The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): The Perspectives of clinical immunologists from China. Clin Immunol 214: 108393.
- Pahan P, Pahan K (2020) Smooth or risky revisit of an old malaria drug for COVID-19? J Neuroimmune Pharmaco 15: 174-180.
- Maurizio Benucci, Arianna Damiani, Maria Infantino, Mariangela Manfredi, Luca Quartuccio (2020) Old and new antirheumatic drugs for the treatment of COVID-19. Joint Bone Spine 87(3): 195.
- Shafieyoon P, Mehdipour E, Mary YS (2019) Synthesis, characterization and biological investigation of glycine-based sulfonamide derivative and its complex: Vibration assignment, HOMO–LUMO analysis, MEP and molecular docking. J Mol Struct 1181: 244-252.
- Shafieyoon P, Mehdipour E, Michalski J (2019) “Synthesis, Characterization, and Biological Investigation of Alanine-Based Sulfonamide Derivative: FT-IR, 1 H NMR Spectra: MEP, HOMO–LUMO Analysis, and Molecular Docking. Russ J Phys Chem A 93 (7): 1285-1296.
- MJ Frisch, GW Trucks, HB Schlegel, GE Scuseria, MA Robb, et al. (2009) Gaussian, Inc., RA Gaussian09, 1, Wallingford CT.
- A Vina (2010) Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading Trott, Oleg; Olson, Arthur J. J Comput Chem 31: 455-461.
- Ruth Huey, Garrett M Morris, Arthur J Olson, David S Goodsell (2007) A semiempirical free energy force field with charge‐based desolvation, J Comput Chem 28: 1145-1152.
- GTM Morris, DS Goodsell, RS Halliday, RH Huey, WE Hart, et al. (1998) Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem 19: 1639-1662.
- AC Wallace, RA Laskowski, JM Thornton (1995) LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Engineering 8: 127-134.
- EE Hodgkin, WG Richards (1987) "Molecular similarity based on electrostatic potential and electric field. Int J Quantum Chem 32: 105-110.
- Paula Jaramillo, Patricia Pérez, Renato Contreras, William Tiznado, Patricio Fuentealba (2006) Definition of a Nucleophilicity Scale. J Phys Chem A 110 (26): 8181-8187.
