 Case Report
Case ReportAbstract
Background: X-linked adrenoleukodystrophy (X-ALD) is a peroxisomal disorder
caused by the accumulation of long chain fatty acids (VLCFA) due to mutations in the
ABCD1 gene.
Case: A 7-year-old boy who started 6 months ago with asthenia, adynamia, loss
of motor skills and decreased school performance. Physical examination showed
hyperpigmented areas in folds, the rest without alterations. Adrenal function was
assessed, finding elevated ACTH (adrenocorticotropic hormone), cortisol was recorded
in normal parameters, due to this, hormonal replacement was started. He presented
an episode of adrenal crisis secondary to community-acquired pneumonia, required
hospitalization and during this hospitalization a brain MRI was performed, which
found a brain pattern compatible with infantile cerebral adrenoleukodystrophy, so he
was sent to the Genetics Department for suspected X-ALD with follow-up in pediatric
endocrinology.
Conclusion: It highlights the importance of making a diagnosis when clinical
suspicion persists, recognizing patterns of the disease with mild to severe manifestations,
depending on the patient, which are used to evaluate the prognosis of the disease, as
well as the importance of VLCFA analysis to promote early diagnosis of ALD.
Keywords: X-Linked Adrenoleukodystrophy; Addison’s Disease; Adrenal Insufficiency; Peroxisomal Disease; Very-Long-Chain Fatty Acid
Abbreviations: X-ALD: X-linked Adrenoleukodystrophy; ACTH: Adrenocorticotropic Hormone; MRI: Magnetic Resonance Imaging; VLCFA: Very Long Chain Fatty Acids; CALD: Cerebral Adrenoleukodystrophy; AMN: Adrenomyeloneuropathy; GTO: Glycerol Trioleate; GTE: Glycerol Trierucate
Introduction
X-linked adrenoleukodystrophy (X-ALD) is the most common peroxisomal disorder, caused by mutations in the ABCD1 locus Xq28 gene due to deficiency of the ALDP protein of the peroxisomal membrane resulting in accumulation of long-chain fatty acids (VLCFA) mainly in the adrenal cortex and central nervous system [1]. It has an incidence of 1 in 21,000 hemizygotes and 1 in 16,800 in heterozygotes. Cerebral infantile X-ALD is the most devastating and progressive phenotype, occurs between three and ten years (peak seven years) is characterized by developmental regression, severe sensory and neurological deficits, in addition to clinical data characteristic of adrenal insufficiency (Addison) [2].
Case Presentation
A 7-year-old boy with no major medical or perinatal history. He started his current condition 6 months ago with asthenia and adynamia, loss of previously learned fine motor skills (buttoning pants, tying shoes), decreased interaction with people and impaired academic performance. Likewise, areas of hyperpigmentation were observed in folds (neck, armpits, elbows, English), so he was requested levels of Adrenocorticotropa hormone ACTH levels of 5455pg/ml (normal: 12 - 76pg/ml) and serum cortisol within normal limits for age. Prednisolone was started at physiological doses. He presented an episode of adrenal crisis secondary to community-acquired pneumonia, so the dose was adjusted to stress doses in the previous 5 months. In this hospitalization, contrasted brain Magnetic Resonance Imaging (MRI) was performed, where hyperintensity was found in the T2 sequence at the level of the white matter in the posterior region of the parietal and occipital lobes, as well as at the level of the midbrain and bridge (Figure 1). He was send to the Genetics department with suspicion of X-linked adrenoleukodystrophy. During his follow-up by Pediatric Endocrinology 2 months after diagnosis, he presented progression of neurological deterioration with ataxic gait, impaired verbal communication and urinary and fecal incontinence. The Genetics department who started management with Lorenzo’s oil (40ml/ day) and lovastatin, as well as a diet low in fatty acids, and requested serum determination of very long chain fatty acids (VLCFA) evaluated him. A follow-up was carried out for 3 months, finding poor response to treatment with persistent cognitive impairment, spasticity and rigidity.
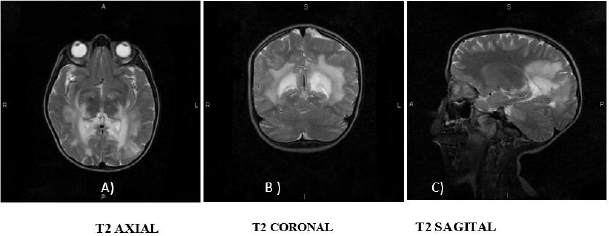
Figure 1: Contrast Brain MRI in T2 sequence.
A. Hyper uptake in the region of the internal capsules and parietal lobes;
B. Hyper uptake is observed in the thalamus, as well as in the white matter of the parietal lobes;
C. Hyper uptake is observed in the corpus callosum, in the bridge and mesencephalon.
Discussion
More than 800 variants in the ABCD1 gene mutation are described, which cause defects in ALDP (adrenoleukodystrophy protein) resulting in the elevation of VLCFA mainly hexadecanoic acids (C26:0) and lignoceric acids (C24:0), causing the accumulation of these in the central nervous system among other tissues [1,2]. VLCFA modify phosphatidylcholine in the myelin membrane can cause instability and inflammation due to altered β-oxidation peroxisomal and increased fatty acid elongation, elevated levels of VLCFA in membranes alter myelin structure and function, this mechanism is described in cerebral adrenoleukodystrophy (CALD) [1]. This causes damage to the endothelium of the cerebral microvasculature critical for the initiation and progression of inflammatory demyelination, tight junction proteins have been shown in autopsies to be displaced, along with massive bloodbrain barrier disruption [1,2]. X-ALD is sensitive to oxidative stress that arises from accumulation of VLCFA and when an excess of free radicals occur they cause the opening of the transition pore of mitochondrial permeability resulting in cell death [1]. Clinical phenotypes of ALD have been described: cerebral infantile, adrenomyeloneuropathy (AMN) and primary adrenal insufficiency [2]. Due to the natural evolution of the disease the phenotypes actually represent a spectrum of the condition, practically all males with ALD will develop adrenal insufficiency and progressive myelopathy in adulthood, and may also develop rapidly progressive cerebral demyelination, which can occur during childhood, but also in adulthood [2].
ALD initially presents as adrenal insufficiency between the ages
of 3-10 years, of these 35-40% may progress to a rapidly progressive
form of inflammatory demyelination (CALD), leading to progressive
neurological deterioration with a peak incidence observed
between the ages of 3-8 years causing a vegetative state until death
within 2-3 years [2,3]. It is clear that other factors modulate this
phenotypic conversion in addition that there is an average delay
in the diagnosis of adrenal insufficiency of 3.5 years, which is
considered a predetermining factor for clinical progression since
an asymptomatic period may be followed [2,3]. The presentation of
the clinical case had a delay in the diagnosis of more than 6 months,
combining the progression of the disease with clinical data such
as asthenia and adynamia in addition to hyperpigmentation at the
same time demonstrating manifestations of adrenal insufficiency,
it is necessary to start the approach by assessing adrenal function
[3]. Adding affectation of fine motor skills involvement suggests
brain magnetic resonance imaging (MRI) to rule out brain tumor,
neuro infection or autoimmune encephalitis [4]. Brain magnetic
resonance imaging (MRI) demonstrates demyelination of the brain
white matter, which usually precedes clinical symptoms, ALD brain
lesions are initially observed in the splenium of the corpus callosum
and the parieto occipital white matter [4,5].
Given the age of the patient, the insidious and progressive onset
together with the episode of adrenal crisis provoked by an infection
in addition to the data provided by the MRI with which a Loes
score of 6 was obtained, it is necessary to approach the patient as a
childhood cerebral phenotype of ALD, requesting VLCFA levels [4,5].
Plasma VLCFA analysis is the most commonly used diagnostic test
for ALD in men [3]. Given the prevalence of 1 in 21,000 newborns
in addition to the difficulty in detecting ADL-X in female patients
due to negative family history with onset between the ages of 30-
40 years plus few clinical data (progressive spastic paraparesis
more common presentation after age 60) and the need for a panel
of laboratory tests [2,3]. In February 2016, newborn screening for
X-ALD was added to the recommended uniform screening panel
in the United States since ALD has been identified as a newborn
screening condition (Figure 2) [2,3]. It was recommended that
asymptomatic males identified by newborn screening in New York
should have their adrenal function quantitatively assessed every
6 months and the first brain MRI performed until 12 months,
then annually until age 3 years, then every 6 months, until age 10
years, and annually thereafter [3]. Once identified, it is suggested
to refer the family for counseling, as well as confirmatory tests in
the members that require them, in addition, the adrenal function is
assessed, since it has been demonstrated that in newborn patients
there is biochemical evidence of adrenal insufficiency from 5 weeks
and at 4.5 months [2,3].
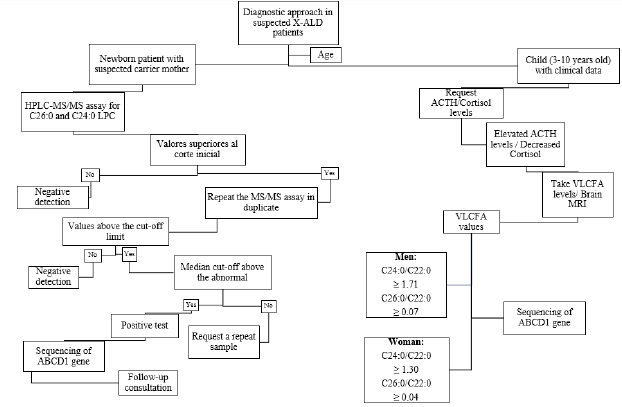
Figure 2: Proposed algorithm for the diagnosis of ALD-X. It is suggested to investigate clinical history in women with clinical data and perform newborn screening [3]. Normal values VLCFA C24:0/C22:0 0.84μg/mL C26:0/C22:0 0.01μg/mL.
Follow-up with brain MRI is suggested to assess disease progression [3,5]. Abnormalities in MRI are evaluated using the Loes scale, points are awarded based on location, extent of brain parenchymal signal changes, ALD patterns, and the presence of focal and global atrophy [5]. Hyperintensity in T1 or T2 may be seen in areas including white matter: supratentorial, corpus callosum, visual pathway, frontopontine or corticospinal tract and major projection fibers [4,5]. The score is rated from 0 (no evidence of disease) to 34 (severe involvement). 5 In conjunction, the neurological function scale that evaluates the clinical progression of the disease in the patient has been used, in our case resulting in a score of 11 (Table 1) [4]. Both scales should be used to assess progression at a minimum of 1 follow-up evaluation [4,5]. CALD is defined as arrested CALD ≥ 2 consecutive MRI scans within a minimum of 6 months with no increase in Loes Score, no contrast enhancement and no progression of brain symptoms (neurological function scale) [4,5]. There is a limitation in the treatment therapies used in patients with ALD, once the diagnosis is made therapeutic interventions are indicated when the Loes score is between 0.5- 9.2.6 Allogeneic hematopoietic stem cell transplantation is effective for cerebral ALD in the early stages of the disease (Loes 0. 5-9 and neurological function score ≤ 1) allowing to stop the disease progression at the brain level, for symptomatic patients with a Loes score ≥ 10 transplantation produces unfavorable results [4,6]. Transplantation does not alter the progression of adrenal insufficiency so corticosteroid replacement should be initiated, hydrocortisone is the glucocorticoid of choice in children and should be started at stress doses as soon as adrenal insufficiency is diagnosed [3].
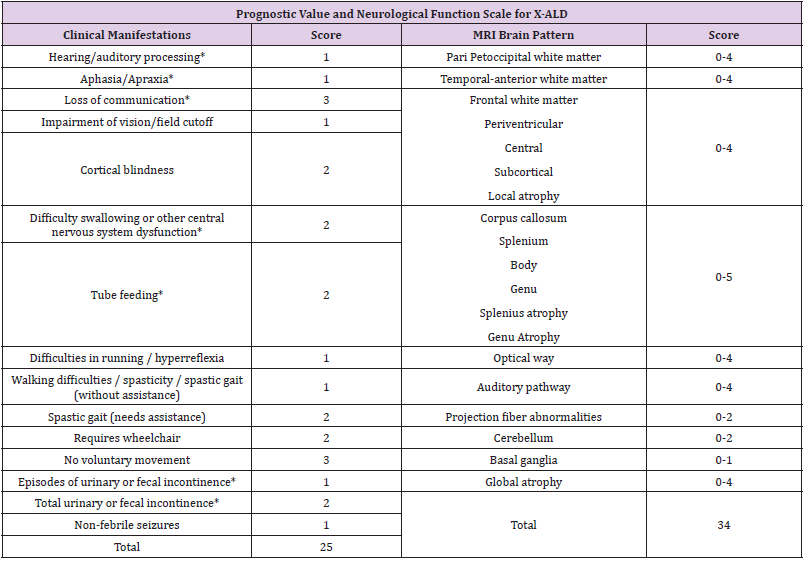
Table 1: Assessment of brain involvement. Neurologic Function Scale is used to evaluate the general clinical neurologic status in patients with CALD to determine the degree of risk prior to stem cell transplantation and post-transplant for follow-up [4]. A score of 0 represents no signs of brain disease but more severe signs (*) represent 12 points that determine a high-risk progression domain [4]. Loes < 10 are considered seriously for allogeneic hematopoietic stem cell transplantation at standard risk. Loes > 10 have a more complicated.
Supportive therapies such as Lorenzo’s oil (GTO/GTE) (mixture of glycerol trioleate [GTO] and glycerol trierucate [GTE] in a ratio 4: 1, at doses 2-3ml/kg/day, this reduces the synthesis of very long chain fatty acids (VLCFA) by competitive inhibition of the enzyme responsible for the elongation of saturated fatty acids, but is ineffective in halting disease progression (CALD). Metabolic modulators such as bezafibrate demonstrated a reduction of VLCFA in ALD fibroblasts, but not in plasma, recently a combination of multiple antioxidants at high doses normalizes biomarkers of oxidative damage and inflammation [2,6].
Conclusion
In this case, the importance of making a diagnosis when clinical suspicion persists, recognizing disease patterns with mild to severe patient-dependent manifestations, which are used to assess disease prognosis, as well as the importance of VLCFA analysis to prompt diagnosis of ALD early to allow hormone replacement and prevent disease progression to a devastating phenotype. MRI images of the brain obtained as part of the patient evaluation are used to determine the use of a specific therapy by assessing the risk-benefit ratio, as well as to evaluate progression and determine if the disease is arrested.
References
- Turk BR, Theda C, Fatemi A, Moser AB (2020) X-linked adrenoleukodystrophy: Pathology, pathophysiology, diagnostic testing, newborn screening and therapies. Int J Dev Neurosci 80(1): 52-72.
- Eng L, Regelmann MO (2020) Adrenoleukodystrophy in the era of newborn screening. Curr Opin Endocrinol Diabetes Obes 27(1): 47-55.
- Regelmann MO, Kamboj MK, Miller BS, Nakamoto JM, Sarafoglou K, et al. (2018) Adrenoleukodystrophy: Guidance for Adrenal Surveillance in Males Identified by Newborn Screen. J Clin Endocrinol Metab 103(11): 4324-4331.
- Miller WP, Mantovani LF, Muzic J, Rykken JB, Gawande RS, et al. (2016) Intensity of MRI Gadolinium Enhancement in Cerebral Adrenoleukodystrophy: A Biomarker for Inflammation and Predictor of Outcome following Transplantation in Higher Risk Patients. AJNR Am J Neuroradiol 37(2): 367-372.
- Loes DJ, Fatemi A, Melhem ER, Gupte N, Bezman L, et al. (2003) Analysis of MRI patterns aids prediction of progression in X-linked adrenoleukodystrophy. Neurology 61(3): 369-374.
- Eichler F, Duncan C, Musolino PL, Orchard PJ, De Oliveira S, et al. (2017) Hematopoietic Stem-Cell Gene Therapy for Cerebral Adrenoleukodystrophy. N Engl J Med 377(17): 1630-1638.
