 Research Article
Research ArticleAbstract
Pharmaceutical companies have now started to reconsider the role of Artificial intelligence (AI) and to develop AI technological competencies for R&D. Drug discovery includes various R&D stages with risk and uncertainty that demand a huge amount of funds. CNS disorder drugs remains the most challenging task in drug discovery. We define the problem of a prototype-driven hypothesis generation as a conditional data generation process for our interesting field. In this article, we share powerful and practical ways that CNS diseases research and development are using AI to improve R&D productivity and to reduce the costs and risks in the pharmaceutical industry. We show the promising result that accelerating drug discovery for CNS disorder and some of these predicted chemical structures accord with novelty and non-obviousness for industrially applicable. This effectively provides a fast and effective drug development method.
Keywords: Artificial Intelligence; FDA; VAE; SMILES; Tau Protein
Abbreviations: AI: Artificial Intel-ligence; QSAR: Quantitative Structure-Activity Relationship; ML: Machine Learning; WIPO: World Intellectual Property Organization; CNS: Central Nervous System; AD: Alzheimer’s Disease
Introduction
Pharmaceutical companies have now started to reconsider the
role of Artificial Intel- ligence (AI) and to develop AI technological
competencies for R&D [1]. Pharma companies are experimenting
with AI in R&D ranging, as a consequence, from in-silico drug
design to clinical trial participant’s identification. The lack of R&D
efficiency and the product gap of the pharma industry coupled
with the yet to be developed AI-competency of drug industry. This
crisis provides great opportunities for AI companies from the core
competence of digital transformation to disrupt the pharma R&D
sector [2]. Quantitative structure-activity relationship (QSAR)
based approaches have proven to be very valuable in predicting
chemical reactivity, physicochemical properties, biological activity,
toxicity, and metabolism of chemical compounds [3]. The lack
of willingness to share proprietary data, the worries about data
quality and the use of AI in decision- making (‘‘garbage-in, garbageout”)
are further hurdles. Pharmaceutical products require longer
and more complex research and development (R&D) cycles than
products in other industries. Consequently, companies invest
significant amounts of money into their new products early on in
their development. New drug development is a business activity
that involves many years of work and major costs. Drug companies
invest significant amounts of money into their new products early
on in their development.
The development of a drug takes 10 to 15 years and was
estimated to cost an average of US $1.2 billion in the early 21st
century, including the costs of failure [4]. These costs have increased
sharply in the last few decades, and pharmaceutical companies
are seeking more efficient approaches to drug development [5].
Drug discovery includes various R&D stages that demand a huge
amount of funds. Moreover, R&D risk and uncertainty including
low efficacy, off-target delivery, time consumption, clinical trials
and regulatory acceptance of the drug in the market could impose a
hurdle and obstacle that impact drug design and discovery pipeline.
The pharmaceutical industry is in a drug-discovery decline.
Changes in the quality of decisions regarding which compound in
drug discovery to take forward are more important than speed
or cost [6]. AI is capable of reducing such high costs in research
and development as it finds solutions faster and with precision.
Artificial intelligence and machine learning technology play a
crucial role in drug discovery and development. Machine Learning
(ML) models and large international data sets offer unprecedented
opportunities to appraise candidate diagnostic, monitoring,
and prognostic markers [7]. There is no doubt that artificial
intelligence is greatly influencing the field of drug discovery. AI
has revolutionized healthcare by dramatically speeding up drug
discovery and development, and thereby brought drug discovery to
patient needs much more quickly.
Life and medical sciences is one of the top three sectors where
AI is most employed, according to the World Intellectual Property
Organization (WIPO). The use of AI in the drug discovery process is
receiving a lot of attention. The main attractions being the promise
of faster, more reliable, and ultimately cheaper development of new
pharmaceutical treatments. How much can AI help hunting for new
drugs with AI for both saving cost and increasing innovation? A key
challenge in drugs candidate screening and advancement of new
chemical entity or new biological entity as therapeutic agents is
accu-rate determination of their drug ability and human toxicity. In
de novo drug design, computational strategies are used to generate
novel molecules with good affinity to the de- sired biological target
[8]. The process of discovering a drug involves many scientists
running thousands of experiments over many years (Figure 1).
However, technological advances in various steps of this process
have not translated into a dramatic increase in FDA-approved
drugs. The vast medical chemical space (comprising >1060
molecules) fosters the development of a large number of drug
molecules. AI – that is including both deep learning and machine
intelligence – has the potential to make waves in drug discovery.
In the past, researchers have attempted to develop expert systems
and knowledge-reasoning systems to solve problems in medical
chemistry. Although AI has had a profound impact on image
recognition areas, comparable advances in drug discovery are rare
[6].

Figure 1: The process of drug discovery, from finding a target to FDA approval. Each step in this process takes years and huge money.
When drug candidates once released to market, innovator
companies recoup their costs in a short period depending on
the blockbuster nature of the product. The quality of decisions
regarding which compound to take forward will have the most
profound overall impact on taking new drugs to market. Healthcare
is just starting to see the impact of AI yet nuclear medicine has been
using intelligent systems including nuclear cardiology, oncology
and neurology on PET/CT for some years [9]. We consider what is
involved and how we should prepare AI deep learning technology
as a competitive advantage for drug discovery of nuclear medicine
(Figure 1). The process of drug discovery, from finding a target to
FDA approval. Each step in this process takes years and huge money.
Drug development often takes a decade of research and billions in
investment before a drug approved by FDA and reaches the market.
This is because the process begins with a millions of chemical
compounds before some candidates are identified for clinical trials.
Artificial Intelligence modeling allows researchers to more quickly
design novel drugs that display the desired drug-like properties.
Researchers could propose synthesizing fewer compounds for
testing fewer experiments in the search for a new candidate.
Designing a new drug is a prolonged and expensive process. The
appeal of AI-de- signed drugs is relatively straightforward. There
are lots and lots of possible molecules that might be useful in
medications, far too many for all the medical researchers in the
world to manually test.
But by using different types of AI, a computer system can come
up with and mine through different molecules, comparing them
against different parameters and learning the most promising
compounds faster than a human could. Drug re- searchers have
used the ‘rule of five’ physical property guidelines for drug
discovery [10]. The application of guidelines linked to the concept
of drug-likeness has gained wide ac-ceptance as an approach to
reduce R&D cost in drug discovery and development process.
However, despite applying in silico models in multi-parameter
optimization of molecules, there is include a greater likelihood of
lack of creativity of hit expansion after target identification for lead
compound identification. In chemistry, as the knowledge available
is often inaccurate, such rules are often ambiguous or even incomplete [11]. On the other hand, there are critical challenges
that ML in chemistry must face, including the detailed description
of chemical space, the flexibility and generalization of models, and
the establishment of effective cross-disciplinary collaborations
[11,12]. Recent evidence from AstraZeneca suggests that a major
reconsideration to research strategy can result in greater success
rates for drug discovery projects reaching lead optimization [13].
After consideration of the potential impact of AI and automated
chemistry, (Table 1) propose five levels of AI-driven drug discovery
to chart the progress of AI rapidly advancing technologies [14].
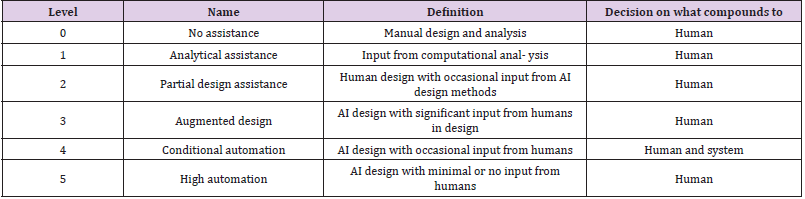
Table 1: Five levels of artificial intelligence-driven drug discovery.
Notably, deep learning-based approaches across compound
design as well as applications of genetic algorithms are highlighted
for medicinal chemistry decision-making [15]. Development and
implementation of deep learning-based methods could considerably
benefit more aspects of early drug discovery. When modeling
the discovery of new compounds as a search problem, we need
an effective representation of the problem. A naive search that
would explore the whole search space of compounds (i.e., in
the order of 1060 states) is not an option. There are constraints
and characteristics for the compounds to discover, such as
substructures of active parts that determine the functions of the
compounds, and chemical core scaffolds that roughly determine
the structures of compounds. These constraints and characteristics
can be given either by a human expert or by Machine Learning AI.
Then, search algorithms should be able to accurately detect the
promising portions of the search space [11]. Why is chemistry
challenging? Among the many compounds available, typically a
very small number meet the “goal state” criteria. It is often difficult
to predict how a com- pound would behave in practice, unless an
experiment is carried out. During this process of prototype-based
drug discovery, an interdisciplinary team of scientists generates
hypothesis about the required changes to the prototype.
Following significant advances in deep learning and related
areas interest in AI has rapidly grown. In particular, the application
of AI in drug discovery provides an opportunity to tackle challenges
that previously have been difficult to solve, such as predicting
properties, designing molecules and optimizing synthetic routes. AI
in Drug Discovery aims to introduce the reader to AI and machine
learning tools and techniques, and to out- line specific challenges
including designing new molecular structures, synthesis planning
and simulation. Furthermore, increasing collaborations among
drug companies to pro- duce patented drugs could also be a major
driver for the industry. AI may reduce a large amount of time
utilized in the drug discovery process. Therefore, reduction in time
factor could be a primary market driver in the forecast period.
Drug development is a slow and increasingly expensive process. AI
has the potential to make the drug development process quicker,
cheaper and more efficient. This technology can make scanning
vast libraries of chemical compounds that might be able to treat
a certain disease easier, can speed up the analysis of biomedical
information from the literature, and can help companies recruit the
most suitable patients for clinical trials. AI can also streamline the
design of better drugs and incorporate new data, such as genomic
analysis, to help personalized medicine become a reality.
The prevalence of Neurological disorders significantly
outnumbers diseases in other therapeutic areas. However,
developing drugs for central nervous system (CNS) disorders
remains the most challenging task in drug discovery, accompanied
with the long timelines and high research costs [16]. The increase
in the incidence of central nervous system disorders such as
Alzheimer’s disease (AD), multiple sclerosis, epilepsy, Parkinson’s
disease and stroke increasing annually, the need for novel
therapeutics to treat CNS disorders has been greater. Although
there is a significant unmet CNS medical need, unfortunately,
because of the low approval rates of drugs targeting those mortal
diseases, that is high risk for R&D spending on CNS disorders [17].
CNS drugs have lower success rates than other drug classes due
to multiple factors, including an insufficient understanding of the
pathophysiology of complex CNS conditions, presence of a blood–
brain barrier (BBB), hard target selection, and the lack of efficacy in
early stages of development [16]. In this article, we share powerful
and practical ways that CNS diseases research and development are
using AI to improve R&D productivity and to reduce the costs and
risks in the pharmaceutical industry.
Materials and Methods
We define the problem of a prototype-driven hypothesis
generation as a conditional data generation process for our
interesting field. Our model is common to start from a molecule,
which already has some of the drug able properties. Such a
molecule, usually called a “prototype” [18], might be selected from
a patent claim or a drug on the market, which could be improved
upon. Within the context of a national patent law, an invention is
patentable if it meets the relevant legal conditions to be granted
a patent. By extension, patentability of new compounds predicted
from AI model also refers to the substantive conditions that must
be met for a patent to be held valid for industrially applicable,
novelty and non-obviousness. The steps of building an AI model
platform for drug discovery (Figure 2). The process for developing
an AI model as follows:
(1) Define the problem appropriately for objective, desired
outputs, etc.,
(2) An interdisciplinary team of scientists generates a hypothesis
about the required changes to the prototype
(3) Prepare the data including collection, exploration and profiling,
SMILES formatting, and improving the data quality,
(4) Use RDKit to select meaningful drug-like features,
(5) Split data into training and validation sets,
(6) Develop and design algorithm for AI model construction,
(7) Train the model through VAE AI model,
(8) Evaluate AI model performance on the validation set and
refine the model,
(9) Evaluate the model on data set of FDA approval drugs data set
not used for method development, and
(10) Change management.
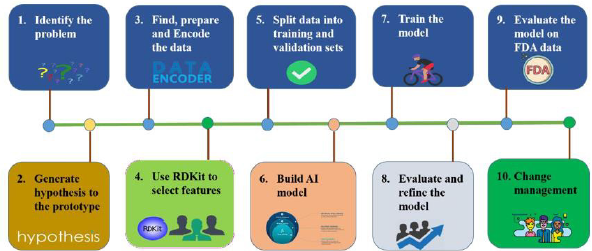
Figure 2: Conceptual framework for our AI model platform assisting drug discovery.
Problem Identification
CNS disorder drugs remains the most challenging task in drug discovery, accompanied with the long timelines and high research costs. Although Tau PET imaging in neurodegenerative tauopathies is still a challenge, but microtubule-associated Tau protein imaging compounds for neurofibrillary tangles is still the promising field for AD. Referred to as tauopathies, some off-target binding led to the optimization of the binding properties and development of secondgeneration of tau tracers. With the aim of developing tracers with better specificity, some of the second-generation tracers were based on the structures of existing tracers (i.e., [18F]RO-948, [18F] PI-2620, [18F]MK-6240, [18F]JNJ311) [19].
Hypothesis Generation
We hypothesized that a constrained generation next to a known prototype [18F]MK- 6240, rather than a no constrained sampling, would yield a better molecule generation. Our model is common to start from a molecule which already has some of the drugable properties. Such prototype molecules might be selected from a patent claim of [18F]MK-6240 (USPTO Pub. No. US 2017/0157274 A1) or a drug on the market which could be im- proved upon. Patentability of new compounds predicted from our AI model to be held valid for industrially applicable, novelty and non-obviousness.
Variational Auto-Eencoder Model
In this issue of Nuclear Medicine for Brain Imaging, we focus on AI for small-molecule- drug discovery purposes, and in this study we take a look at how AI is being utilized throughout the drug design and development processes, and the positives and negatives posed by our AI technology. Our Variational Auto Encoder (VAE) [20] algorithm uses a python-based AI system to find a suitable candidate in drug discovery. The (Figure 3) shows VAE architecture for our brain imaging AI model. The (Table 2) shows the hyper parameters for our VAE model. VAE starts by encoding the molecule in simplified molecular-input line-entry system (SMILES) representation from the ZINC database using the encoder function, then applying convolutions over various filter sizes that correspond to chemical substructures. During generation, the vectors of features are sampled from the prior distribution and their output is passed to the decoder that generates a new representation. VAE uses simple representation of molecules by their SMILES strings only. Generative models are trained with a stack-augmented memory network to produce chemically feasible SMILES strings, and predictive models are derived to forecast the desired novel de novo–generated compounds.
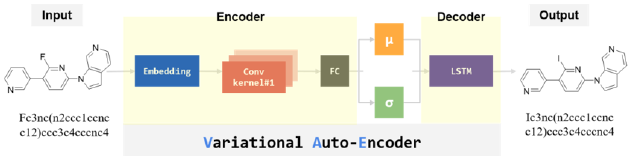
Figure 3: Varitional Auto-Encoder for our AI model.
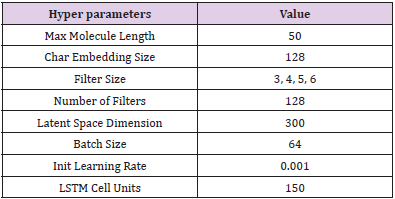
Table 2: Hyper parameters for Varitional Auto-Encoder Model.
Data Preparation
We prepare the data including collection, exploration and
profiling, chemical structure formatting, and improving the data
quality. To connect chemistry with AI language, it is important to
understand how molecules are represented. Usually, in molecular
graphs in models for natural language processing, the input and
output of the model are usually sequences of single letters, strings
or words. We therefore employ the SMILES format, which encodes
molecular graphs compactly as human-readable strings. SMILES is
a formal grammar which describes molecules with an alphabet of
characters, for example c and C for aromatic and aliphatic carbon
atoms, O for oxygen, -, = and # for single, double and triple bonds.
To indicate rings, a number is introduced at the two atoms where
the ring is closed. For example, toluene in aromatic SMILES notation
would be Cc1ccccc1. Side chains are denoted by round brackets. To
generate valid SMILES, the generative model would have to learn
the SMILES language models [8,21]. This VAE model had trained on
a subset with approximately 250k drug-like com- pounds extracted
from ZINC database (a free tool to discover chemistry for biology)
randomly [22]. In order to make the resulting compounds more
novel, we had added a layer of diversity before the decoding layer
to produce similar but different structures.
In addition, the databases have subsets of ZINC filtered by
physical properties. One such filtering is based on Lipinski’s rule
of five, which is a heuristic method to evaluate if a molecule can
be a drug candidate. Our VAE model was trained on a subset with
approximately 200k drug-like compounds extracted at random from
the ZINC drug-like data- base. The subset was further divided into
train/validation/test sets, with 50k compounds for the validation
and test sets, and the rest for the training set. The subsets were
used for training the model (train), evaluating hyper parameters
and stopping criteria (validation), and for method evaluation and
experiments (test). As simple drug-like properties, the molecular
weight (MW), number of heavy atoms, number of rotatable bonds,
number of aromatic rings, number of chiral centers, lipophilicity
e.g., octanol-water partition coefficient (logP), polarity [e.g., polar
surface area (PSA), and number of hydrogen bond donors (HBD)
and hydrogen bond acceptors (HBA) are the main developmental
parameters analyzed during the lead to clinical candidate pro- cess.
The (Figure 4) shows python programming logic on Lipinski’s rule
of five for VAE model.

Figure 4: Filtering is based on Lipinski’s rule of five for VAE model.
Results and Discussion
This project uses 250,000 Drug-Like database as the training
set of artificial intelligence model. Before starting the training,
confirm the reliability of the database by analyzing and statistically
whether the data in the database meets the Gaussian distribution.
The result is shown in (Figure 5). After confirming that the data
is Gaussian, the database is a reliable database, and the molecular
weight distribution is 150 to 500. Dalton (Da); the distribution
coefficient (log P) ranges from -3 to +6. The larger the value, the
higher the lipophilicity of the substance to the organic phase and
the lower the hydrophilicity. The log P of non-polar compounds
is generally greater than zero, and the log P of polar com- pounds
is generally less than zero. This 250,000 Drug-Like database is a
database that conforms to the Gaussian distribution and can be
used as a training set for artificial intel- ligence. Our database has
been used for calculating the five target properties (MW, LogP, HBO,
HBA, rotatable bonds) and validating the generated SMILES codes
of the molecules for CNS drug discovery. The total dataset is made
of molecules randomly selected from the ZINC dataset. In the end of
training, our model has reached 97.68% accuracy. In order to test
the predictive ability of the artificial intelligence model for drug innovation,
we use the FDA-approved and public drug list as the input
of the model to develop new drugs.
Using FDA-approved drugs as input molecules for prediction,
the artificial intelligence model will use the above-mentioned
drugs as the center point of the compound vector space and look
for potential compounds around the center point. Here we make a
hypothesis: In the compound vector space, neighboring compounds
should have similar properties, so the aforementioned effective
results predicted by FDA drugs should have similar properties to
the original drugs. After the output results, our model can take FDA
drugs as input and predict another known FDA drug. For example,
with Pheneilzine as input, the model predicts Isoniazid as shown in
(Table 3). It is worth mentioning that the above two neither input
nor output appears in the training set. In addition, this project also
attempts to use another method for model validation. Input 130
MK-6240 analogue structures into the VAE artificial intelligence
model completed by 250,000 Drug-like trainings to predict new
molecules. The new molecules produced are verified by RDKit to
see if they meet the gold standard for drugs. A total of 28 molecules
meet According to the gold standard of drugs, 7 compounds
(INER-M-1~7) are screened out by drug development researchers
to evaluate novelty as shown in (Table 4). The input data of the 7
compounds with inhibition constant (Ki) screened are shown in
(Table 5) (INER-1~7).
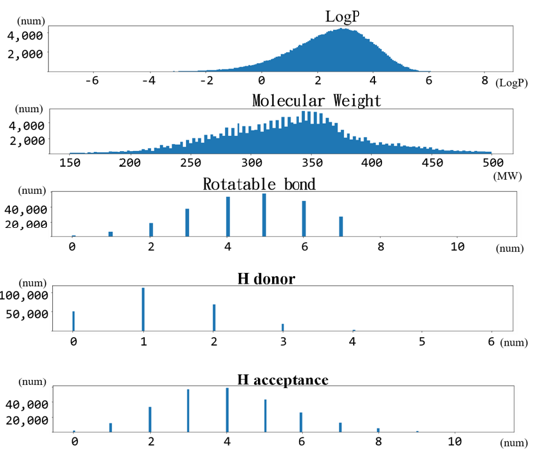
Figure 5: This 250,000 Drug-Like database database meets the Gaussian distribution including the parameters of distribution coefficient (log P), molecular weight distribution, rotatable bond, H proton donor and H proton acceptance.

Table 3: Use a FDA drug Phenelzine Served as a Prototype to the Generation Process.
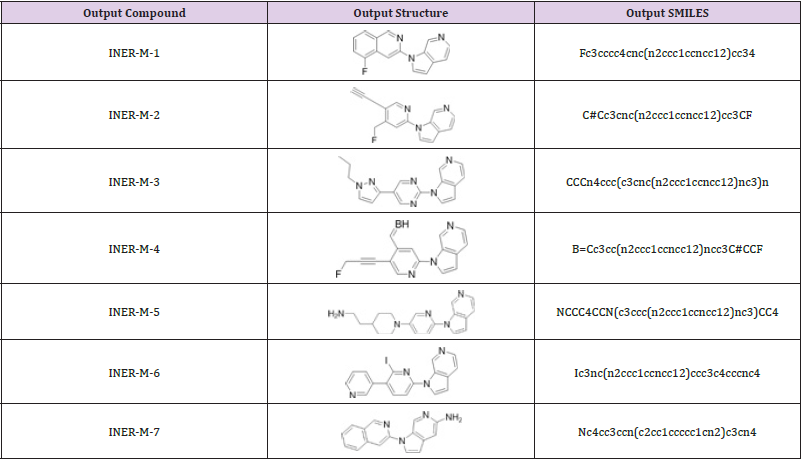
Table 4: Small molecule structures produced by artificial intelligence models (INER-M-1~7).
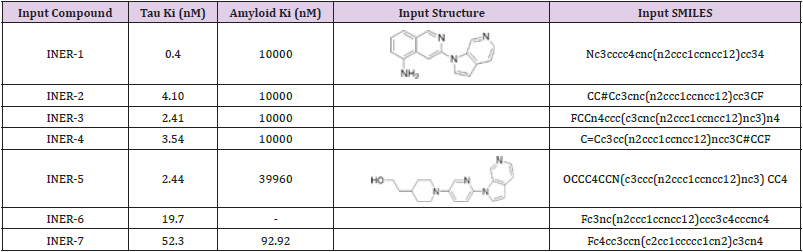
Table 5: Artificial intelligence model input data (INER-1~7).
Four binding sites were identified for the PET radiotracers MK-6240 [23]. We use molecular docking simulation to predict the interaction energy score of MK-6240 and AI design compounds with Tau fibril and analyze its binding force with Tau fibril. Computer simulation (Software: Discovery Studio 2020, DS 2020) of small molecule structures (INER-M-1~7) produced by artificial intelligence, and molecular docking analysis on four binding sites on Tau fibril, Tau fibril protein structure and junction sites are shown in (Figure 6), and the results of docking analysis of artificial intelligence compounds are shown in (Table 5). Knowing the location of the binding site prior to docking will in- crease the docking efficiency. If the brain nerve drug MK-6240 is used as the standard, at each binding site, at least 4 compounds produced by artificial intelligence are better than MK-6240, indicating that the artificial intelligence model developed in this project has the opportunity to predict more docking advantage of small molecule compounds. We use the 3D surface view of paired helical filament in Alzheimer’s disease brain PDB code:5O3L from Protein Data Bank (PDB) for docking simulations.
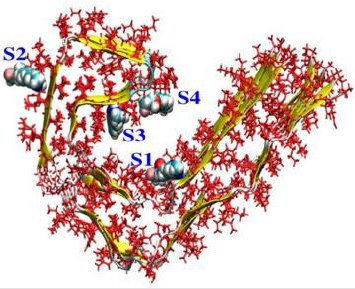
Figure 6: Chimera AD Tau model showing 4 various high-affinity binding sites of tau protofibril. The site 2 is termed a surface site as it is exposed to a greater amount of solvent molecules. The sites 1, 3, and 4 are termed core sites as they are buried inside the fibril [23,24].
We used the DS 2020 software to evaluate the interaction energies of INER-M-1~7 at these four binding sites and compared them with MK-6240. For MK-6240, the lowest energies were found for Site 1, while for INER-M-3, INER-M-5 and INER-M-6 were more interaction energies for the Sites 1, 2, 3 and 4 (Table 6). For docking research, we use the INTERACTION ENERGY as the basis for judging docking scores. Among the 8 design derivatives, except for compound 4, all of them can be successfully docked to the four binding sites of Tau fibril. Among them, compound 5 has the largest INTERACTION ENERGY at all the four binding sites, indicating that the binding to Tau protein is relatively stable, and compared with the lead compound MK- 6240, compounds 5, 3, and 6 all have larger INTERACTION ENERGY at the four binding sites. For compound 5, compound 5 has the largest number of four binding sites INTERACTION ENERGY. The docking score at Site2 is higher than the other three binding sites, reaching 61.03, followed by Site4, Site3, and Site1. The analysis of the force of compound 5 at Site2 is shown in (Figures 7 & 8). The most important force is that the pyridine of compound 5 has charge interaction with GLU33 and Pi interaction with LYS35. The (Table 7) shows the notation of our figures for amino acids. For compound 3, the docking score of compound 3 at Site2 was higher than the other three binding sites, reaching 51.54, followed by Site4, Site3, Site1 [24].
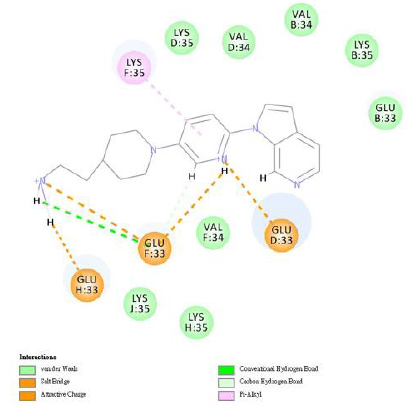
Figure 7: The analysis of the interaction force of compound 5 at Site2. Compound 5 forms one van der Waals force and multiple charge interactions with the active site residues of PDB code 5O3L.
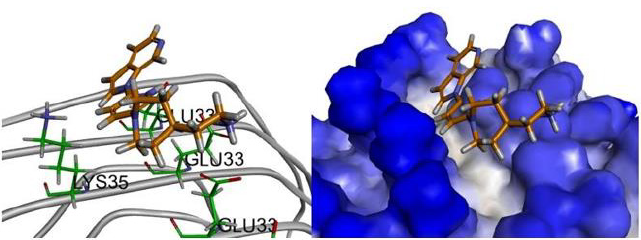
Figure 8: The compound 5 conformation images, the putative binding mode of compound 5 in the 3D surface view of paired helical filament in Alzheimer’s disease brain (PDB code: 5O3L) derived from docking simulations.
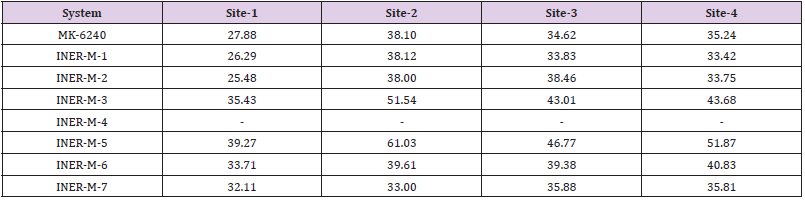
Table 6: Calculated Interaction Energies (kcal/mol) from Molecular Docking Studies for Various Tracers in Four*.
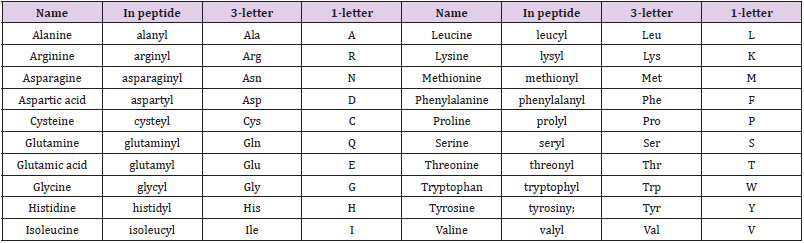
Table 7: Notation for amino acids*.
The force analysis of compound 3 at Site2 is shown in (Figures 9 & 10). The pyridine of compound 3 has charge interaction with GLU33, Pi interaction with LYS35, and H Bond interaction with GLU33 and VAL34. For compound 6, the docking score of compound 6 at Site4 was higher than the other three binding sites, reaching 40.83, followed by Site2, Site3, Site1. The analysis of the force of compound 6 at Site4 is shown in is shown in (Figures 11-13). The most important force is that the pyridine of compound 6 has Charge interaction and Pi(π) interaction with LYS48.
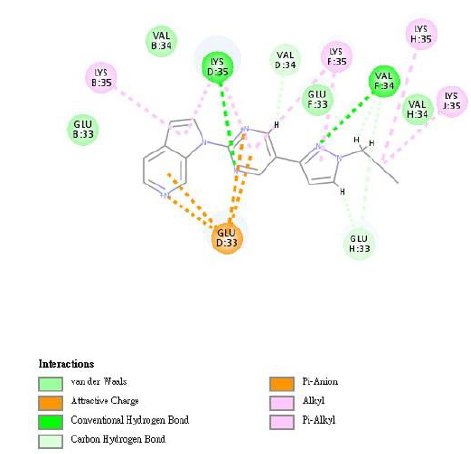
Figure 9: The analysis of the interaction force of compound 3 at Site2. Compound 3 forms multi-ple force and multiple charge interactions with the active site residues of PDB code 5O3L.
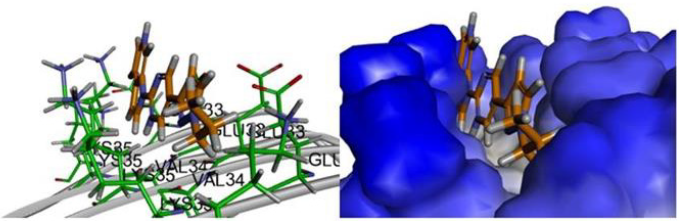
Figure 10: The analysis of the interaction force of compound 3 at Site2. Compound 3 forms multi-ple force and multiple charge interactions with the active site residues of PDB code 5O3L.
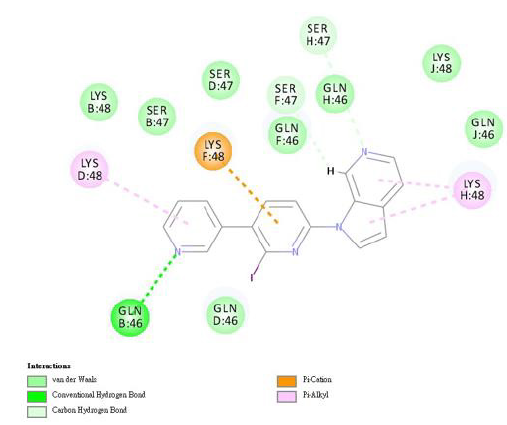
Figure 11: The analysis of the interaction force of compound 3 at Site2. Compound 3 forms multi-ple force and multiple charge interactions with the active site residues of PDB code 5O3L.
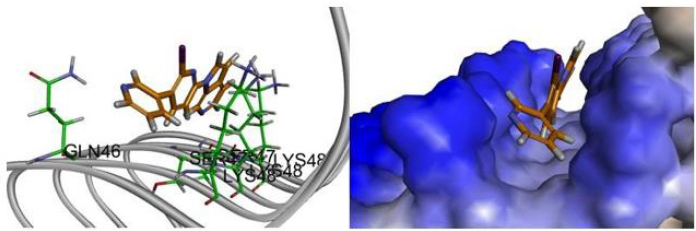
Figure 12: The compound 6 conformation images, the putative binding mode of compound 6 in the 3D surface view of paired helical filament in Alzheimer’s disease brain (PDB code: 5O3L) de- rived from docking simulations.
Figure 13: AI/ML applications in the drug discovery pipeline.
Conclusion
We argue that medical chemistry should be the next grand challenge for Artificial Intelligence. We proposed a new molecular design strategy based on the variational auto-encoder. Instead of high-throughput virtual screening, our method as one of the deep learning-based generative models directly produces molecules with desirable target properties. In particular, its strength is controlling multiple target properties simultaneously by imposing them on a condition vector. AI in drug discovery will need to collaborate the scientists and the scientific method. The AI process of drug discovery consisting of identifying questions, generating testable hypotheses, proving hypotheses, collecting and analyzing data, testing favorable pharmacokinetics in mice and drawing conclusions that lead to new paradigm. We show the promising result that accelerating drug discovery for CNS disorder, and some of these predicted chemical structures accord with novelty and non-obvious- ness for industrially applicable. This effectively provides a fast and effective drug development method. However, a lot of research remains to be done and this paper is outlining how a focused, good hypotheses and collaborative effort between AI and drug research scientists could produce valuable discoveries and contributions to medical science for the benefit of humanity.
Author Contributions
Conceptualization, M.-H.L and S.-J.Y.; methodology, M.-H.L and S.-J.Y.; software, L.-H.H.; validation, L.-H.H. and K.-H.C.; resources, L.- H.H., K.-H.C. and S.-W.L.; data curation, L.-H.H. and K.- H.C.; writing— original draft preparation, M.-H.L; writing—review and editing, M.-H.L. and S.- W.L; supervision, L.-H.H.; project administration, M.- H.L. and S.-J.Y.; All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Institute of Nuclear Energy Research, Taiwan.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Schuhmacher A, Gassmann O, Kuss M, Hinder M (2019) The Art of Virtualizing Pharma R&D. Drug discovery today 24: 2105-2107.
- Kim JA (2013) Creative Destruction of Medicine: How the Digital Revolution Will Create Better Health Care. Healthcare Informatics Research 19: 229.
- Neves BJ, Braga RC, Melo Filho CC, Moreira Filho JT, Muratov EN, et al. (2018) QSAR-Based Virtual Screening: Advances and Applications in Drug Discovery. Frontiers in pharmacology 9: 1275.
- Swarbrick B (2014) The Current State of near Infrared Spectroscopy Application in the Pharmaceutical Industry. Journal of Near Infrared Spectroscopy 22: 153-156.
- Takahashi K, Sengoku S, Kimura H (2011) Driving clinical study efficiency by using a productivity breakdown model: Comparative evaluation of a global clinical study and a similar Japanese study. J Clin Pharm Ther 36(1): 87-98.
- Bender A, Cortes-Ciriano I (2020) Artificial intelligence in drug discovery: what is realistic, what are illusions? Part 1: Ways to make an impact, and why we are not there yet. Drug discovery today 26(2): 511-524.
- Grollemund V, Pradat PF, Querin G, Delbot F, Le Chat G, et al. (2019) Machine Learning in Amyotrophic Lateral Sclerosis: Achievements, Pitfalls, and Future Directions. Frontiers in neuroscience 13: 135.
- Segler MHS, Kogej T, Tyrchan C, Waller MP (2018) Generating Focused Molecule Libraries for Drug Discovery with Recurrent Neural Networks. ACS central science 4(1): 120-131.
- Hall M (2019) Artificial intelligence and nuclear medicine. Nucl Med Commun 40: 1-2.
- Lipinski CA (2004) Lead- and drug-like compounds: the rule-of-five revolution. Drug discovery today. Technologies 1: 337- 341.
- Kishimoto A, Buesser B, Botea A (2018) AI Meets Chemistry. AI Meets Chemistry. Proceedings of the AAAI Conference on Artificial Intelligence 32(1).
- Gentile F, Agrawal V, Hsing M, Ton AT, Ban F, et al. (2020) Deep Docking: A Deep Learning Platform for Augmentation of Structure Based Drug Discovery. ACS central science 6: 939-949.
- Morgan P, Brown DG, Lennard S, Anderton MJ, Barrett JC, et al. (2018) Impact of a five-dimensional framework on R&D productivity at AstraZeneca. Nature reviews. Drug discovery 17: 167-181.
- Clive P Green, Ola Engkvist, Garry Pairaudeau (2018) The convergence of artificial intelligence and chemistry for improved drug discovery. Future Medicinal Chemistry 10(22).
- Panteleev J, Gao H, Jia L (2018) Recent applications of machine learning in medicinal chemistry. Bioorg Med Chem Lett 28: 2807-2815.
- Vatansever S, Schlessinger A, Wacker D, Kaniskan HU, Jin J, et al. (2021) Artificial intelligence and mach ine learning-aided drug discovery in central nervous system diseases: State-of-the-arts and future directions. Medicinal research reviews 41: 1427-1473.
- Alzheimer’s and Dementia Research.
- Harel S, Radinsky K (2018) Accelerating Prototype-Based Drug Discovery using Conditional Diversity Networks. Association for Computing Machinery, pp. 331-339.
- Leuzy A, Chiotis K, Lemoine L, Gillberg PG, Almkvist O, et al. (2019) Tau PET imaging in neurodegenerative tauopathies-still a challenge. Molecular psychiatry 24: 1112-1134.
- Diederik K, Welling M (2014) Auto-Encoding Variational Bayes. Conference: ICLR.
- Popova M, Isayev O, Tropsha A (2018) Deep reinforcement learning for de novo drug design. Science Advances 4(7).
- Sterling T, Irwin JJ (2015) ZINC 15--Ligand Discovery for Everyone. Journal of chemical information and modeling 55(11): 2324-2337.
- Murugan NA, Nordberg A, Agren H (2018) Different Positron Emission Tomography Tau Tracers Bind to Multiple Binding Sites on the Tau Fibril: Insight from Computational Modeling. ACS chemical neuroscience 9: 1757-1767.
- Mukherjee J, Liang C, Patel KK, Lam PQ, Mondal R (2021) Development and evaluation of [(125) I]IPPI for Tau imaging in postmortem human Alzheimer's disease brain. Synapse 75(1): e22183.
