 Short Communication
Short CommunicationAbstract
Background: Rett syndrome is a neurodegenerative disorder with a period of developmental regression and deceleration of brain growth after a relatively normal neonatal course. The hallmark of Rett syndrome is repetitive handwringing. The main criteria include gait abnormalities and stereotypical hand movements (hand wringing and squeezing, clapping and tapping, hand-to-mouth and object-to-mouth activities, washing, and rubbing), and losses of acquired purposeful hand skills and of acquired spoken language. Atypical Rett syndrome, however, meets at least two main criteria. A magnetic resonance image (MRI) in a Rett syndrome patient is usually not specific. The genetic causes include MECP2, CDKl5, and FOXG1.
Case Presentation: We have presented a case of atypical Rett syndrome with three of the four main criteria. Genetic examinations of candidate genes of MeCP2, CDKL5 and FOXG1 were negative. The brain MRI showed periventricular nodular heterotopia.
Conclusions: We recommend an MRI study when Rett syndrome is suspected
Abbreviations: OMIM: Online Mendelian Inheritance in Man; EEG: Electroencephalogram; WES: Whole-Exome Sequencing; MRI: Magnetic Resonance Image; ABAS-II: Adaptive Behavior Assessment System®-Second Edition; NCBI ClinVar: National Center for Biotechnology Information, clinical variability and predictability
Introduction
Rett syndrome (OMIM # 312750) is a neurodegenerative disorder with a period of developmental regression and deceleration of brain growth after a relatively normal neonatal course. It was first described in 1966 [1]. Its incidence rate is approximately 1 in 15,000-22,000 children, and it occurs predominantly in girls. Rett syndrome is most commonly caused by mutations in MeCP2, a transcription factor that binds to methylated CpG islands and silences transcription. Other genetic causes include CDKL5 and FOXG1 [2]. The hallmark of Rett syndrome is repetitive handwringing [3]. All patients have autism spectrum disorder. After a relatively normal neonatal period, acquired microcephaly and the regression of purposeful hand skills and spoken language become apparent. Additional symptoms include generalized tonicclonic convulsions, breathing abnormalities with peculiar sighing respirations, intermittent apnea, feeding disorders, and poor weight gain [3]. There is no specific therapy for Rett syndrome. Generally, girls survive to adulthood.
Patient History
A 5-year-old girl was brought to our outpatient department because she had not spoken for 2 years. She was healthy except for her repetitive hand-wringing and mild developmental delay of gross motor skills. She did not speak, even after speech therapy. Her eye contact was poor, but her response to sound was good. Her head circumstance was less than 3 percentile. Physical and neurological examinations showed no specific abnormalities (i.e., no muscle rigidity or ataxic gait). Laboratory data were: white blood cell count = 9.42 × 109/L; hematocrit = 38.6%; platelet count = 371 × 109/L; and creatinine kinase = 218 U/L. Brainstem auditory evoked potential were normal. Her electroencephalograms (EEG) showed unremarkable finding. The Adaptive Behavior Assessment System®-Second Edition (ABAS-II) including three major areas of functioning assessed Conceptual Reasoning, Social Interactions, and Practical Functioning were all very low to 40 scores by a psychologist evaluation. Brain magnetic resonance imaging (MRI) showed two nodular structures (< 1 cm long) in her bilateral periventricular white matter (Figure 1, arrows) and suspicious periventricular nodular heterotopia. She was diagnosed with atypical Rett syndrome. Genetic examinations of MeCP2,CDKL5 and FOXG1were negative.
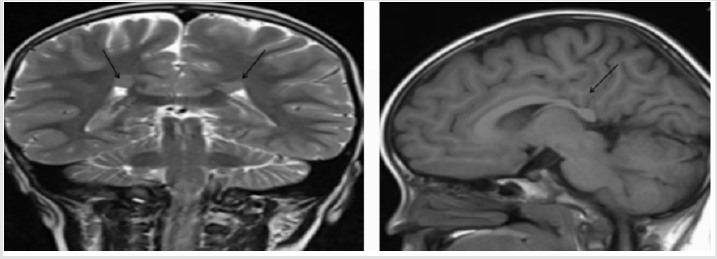
Figure 1:Brain MRI coronal view (left) and sagittal view (right) shows nodular structures (black arrows) in bilateral periventricular white matter. Periventricular nodular heterotopia was diagnosed./p>
Discussion
Rett syndrome is divided into four stages. Stage 1 is early onset: 6-18 months old. The child has less eye contact, less interest in toys, and delayed gross motor skills. Stage 2 (1-4 years old) is called “rapid destruction,” and is characterized by losses of acquired purposeful hand skills and acquired spoken language, which are replaced with stereotypical hand movements. Stage 3 (2-10 years old) is called “plateau.” It can persist for years, during which hand use and communication skills improve. Stage 3 (> 10 years old) is called “late motor deterioration,” which can persist for decades [4]. It is characterized by reduced mobility, increased rigidity, and spasticity. Some children in this stage are unable to walk. A diagnosis of Rett syndrome requires all main criteria [4]. Moreover, brain injury secondary to trauma, neurometabolic disease or severe infection, and grossly abnormal psychomotor development in first 6 months of life should be excluded [4]. The main criteria include gait abnormalities and stereotypical hand movements (hand wringing and squeezing, clapping and tapping, hand-to-mouth and object-to-mouth activities [mouthing], washing and rubbing) and losses of acquired purposeful hand skills and of acquired spoken language. Atypical Rett syndrome, however, meets at least two main criteria and at least five supportive criteria: breathing disturbances, bruxism, impaired sleep pattern, abnormal muscle tone, peripheral vasomotor disturbances, scoliosis and kyphosis, growth retardation, small cold hands and feet, inappropriate laughing and screaming spells, intense staring, and a diminished response to pain. MeCP2 mutations have been detected in approximately 92% of typical cases [1,5]. There are three specific variant forms of atypical Rett syndrome: preserved speech variant, early seizure variant, and congenital variant [4]. Mutations in MeCP2 are associated with the preserved speech variant. However, mutations in CDKL5 and FOXG1 have been discovered in the early seizure variant and the congenital variant, respectively. TUBB2B mutation, p.Ala248Val, has been reported previously as a de novo variant in two unrelated individuals with polymicrogyria, intellectual disability, and epilepsy [6,7]. In summary, we have presented a case of atypical Rett syndrome with three of the four main criteria. Genetic examinations of MeCP2, CDKL5and FOXG1 were negative. The brain MRI showed periventricular nodular heterotopia.
References
- Rett A (1966) On a unusual brain atrophy syndrome in hyperammonemia in childhood. Wien Med Wochenschr 116(3): 723-6.
- Ehrhart F, Sangani NB, Curfs LM (2018) Current developments in the genetics of Rett and Rett-like syndrome. Curr Opin Psychiatry 31(2): 103-108.
- Hagberg B, Witt Engerström I (1986) Rett syndrome: a suggested staging system for describing impairment profile with increasing age towards adolescence. Am J Med Genet Suppl 1: 47-59.
- Neul JL, Kaufmann WE, Glaze DG, Christodoulou J, Clarke AJ, et al. (2010) Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol 68(6): 944-50
- Percy AK, Lane JB, Childers J, Skinner S, Annese F, et al. (2007) Rett syndrome: north American database. J Child Neurol 22(12): 1338-41.
- Stottmann RW, Donlin M, Hafner A, Bernard A, Sinclair DA, et al. (2013) A mutation in Tubb2b, a human polymicrogyria gene, leads to lethality and abnormal cortical development in the mouse. Hum Mol Genet 22(20): 4053-63.
- Amrom D, Tanyalçin I, Verhelst H, Deconinck N, Brouhard GJ, et al. (2014) Polymicrogyria with dysmorphic basal ganglia? Think tubulin! Clin Genet 85(2): 178-83.
