 Review Article
Review ArticleAbstract
Disorders of sex devolvement (DSD), formerly, known as ambiguous genitalia or intersex disorders, represent a spectrum of congenital conditions in which the chromosomal, gonadal or anatomic sex is atypical. DSD are relatively rare disorders with diverse etiologies. The estimated prevalence is approximately 0.1 to 0.2% world-wide, i.e, 1 in 5000 live birth. It constituted a major complex social and medical emergency that requires a multidisciplinary coordinated team of specialists. DSD is vary in frequency with congenital adrenal hyperplasia (CAH) is being the commonest. Several forms can lead to wrong sex assignment with its consequences, as well it can lead to salt loss which if unrecognized early and not appropriately treated can lead to salt wasting, dehydration and adrenal crisis. The team consists of pediatric endocrinologist, pediatric radiologist, geneticist, pediatric surgeon, urologist, plastic surgeon, child psychologist or psychiatrist, and other specialties such as a nurse and gynecologist to be consulted whenever needed. The team should be involved from an early stage. The first step in management is to assign the correct sex of rearing in a timely manner, to avoid the issue of sex-reassignment and its devastating consequences. It should be based on multiple factors, such as genotype, the presence, location and appearance of the reproductive organs. The pediatric radiologist plays a significant and important role in elucidating existing anatomy by utilizing the various radiological tools. Great advances have been made over recent years in the diagnosis and management of DSD, most notably in the field of molecular genetics technologies which have advanced our understanding of the disorder. Furthermore, DSD surgery which is more challenging has been entirely changed. Family dynamic and sensitivity of the issue play an essential role in the care of individual with DSD. Finally, assessment of family coping skills and support systems are essential. The main focus of this article is to understand the various etiologies of DSD, diagnostic approach, and management of infants and children with suspected disorders of sex development (DSD) are discussed.
Keywords: Ambiguous Genitalia; Aetiology; Disorders of Sex Development (DSD); Diagnosis; Genitogram; Endocrine Assessment; Laparoscopy; Magnetic Resonance Imaging (MRI); Treatment; Ultrasound; Surgery; Sex Assignment
Introduction
Ambiguous genitalia is a Latin term, which indicates a condition in which the external genital organs do not have the appearance typical of either a male or female (Figures 1-3). It is still confusing to both parents, patients and doctors. As a consequences international expert in pediatric endocrinology met at Chicago, under the auspices of the European Society for Pediatric Endocrinology and the Lawsn-Wilkins Pediatric Endocrinology (U.S.A), where they suggested the current definition of disorders of sex development (DSD) as a congenital condition in which development of chromosomal, gonadal, or anatomical sex is atypical. It is clear and understandable for doctors and patients, as well as it covers all aspects of the disorders in sex development. It is not that rare, and constitute a complex, major social and medical emergency. DSD is vary in frequency with Congenital Adrenal hypoplasia (CAH) is the common of ambiguous genitalia in the newborn worldwide. Several forms of congenital adrenal hyperplasia can lead to significant salt loss, which, if unrecognized and not appropriately treated, may lead to salt wasting, dehydration and adrenal crisis. To ensure that the affected individual has a high quality of life (a successful outcome), medical practitioners must quickly and correctly assign the individual’s gender in a timely manner with the help of a multidisciplinary coordinated team of specialists. The team consists of a pediatric endocrinologist, pediatric radiologist, geneticist, pediatric surgeon, urologist, plastic surgeon, child psychologist or psychiatrist, and other specialties such as a nurse and gynecologist to be consulted whenever needed. The team should be involved from an early stage.

Figure 1: Ambiguous genitalia in a 46, XX patient known to have congenital adrenal hyperplasia (CAH) due to 21α-hydroxylase deficiency. Note the complete masculinisation, with normal looking hyperpigmented male genitalia. (But no palpable testes).

Figure 2: Ambiguous genitalia in a 46, XY patient known to have congenital adrenal hyperplasia (CAH) due to 3β-hydroxysteroid dehydrogenase (HSD) deficiency. Note the pigmented, short, curved phallus, central urogenital slit, and separated labioscrotal folds with palpable gonads.

Figure 3: Ambiguous genitalia in a 46 XY with the diagnosis of complete androgen insensitivity (testicular feminization) syndrome. Note the two palpable gonads (testes) within the labioscrotal fold and urogenital slit.
When approaching the treatment of any child diagnosed with sexual ambiguity, it is important to first review and understand the embryology and physiology of sexual differentiation. Although the basic developmental events have long been known, the genetic, biochemical, endocrine, and molecular mechanisms are complex and have only been partially elucidated. Whereas surgical reconstruction of the genitalia has been widely applied in infants and children with DSD in the past, the growing understanding of the psychological and social implications of gender assignment has showed the paradigm away from early surgery in some affected patients [1-12]. The main focus of this article is to understand the various etiologies of DSD. Diagnostic approach and management of infants and children with suspected disorders of sex development (DSD) are discussed.
The Etiological Causes of DSD
A major step involved in the management of patients with disorders of sex development (DSD) and ambiguous genitalia is to understand the various etiological causes, Table 1 and Table 2. Great advances have been made over recent years in the diagnosis and management of DSD, most notably in the field of molecular genetics technologies which have uncovered, more novel molecular etiologies. Congenital Adrenal Hyperplasia (CAH) is the most common of ambiguous genitalia in the newborns worldwide, while mixed gonadal dysgenesis (MGD) is the second most frequent cause [13,14].
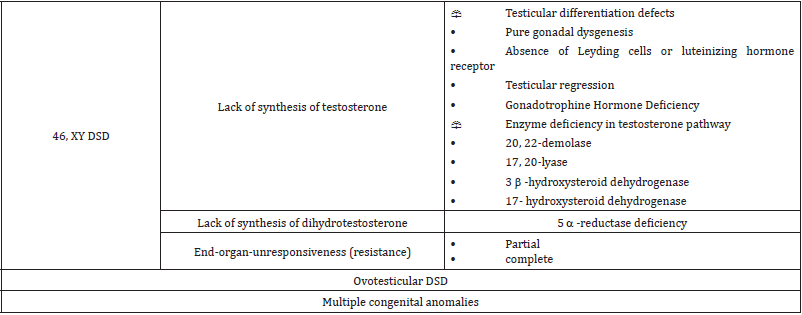
Table 1: Major causes of disorder of sex development (DSD) in patients with 46XY karyotype.
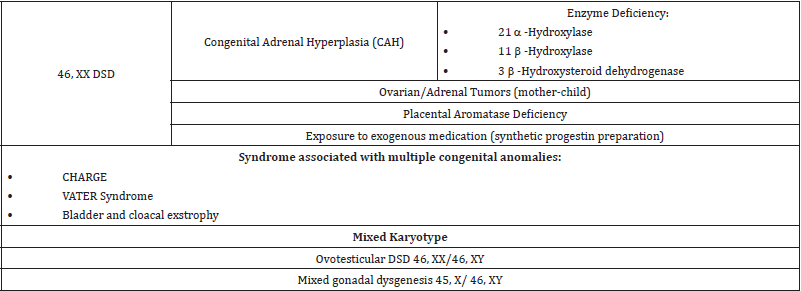
Table 2: Major causes of disorders of sex development (DSD) in patient with 46 XX, and mixed karyotypes.
Diagnostic Approach of the Disorders of Sex Development (DSD), and the Issue of Sex Assignmen (Sex of Rearing)
The main goal of the management is to reach a correct diagnosis, in a timely manner, to avoid the issue of sex-reassignment and its consequences. Patients with disorders of sex development (DSD), and ambiguous genitalia, should be managed in a highly specialized medical center, where a multi-disciplinary team of specialists capable of managing such disorder is present. The team consists of pediatric endocrinologist, geneticist, pediatric surgeon, urologist, plastic surgeon, child psychologist or psychiatrist, pediatric radiologist, social worker and other specialties such as a nurse and gynecologist to be consulted whenever needed. The team should develop and outline a clear plan for the clinical management. Parents need to be supported as they are shocked and require counselling. The team should provide parents with the initial information clearly and openly and advise them to delay the registration of the birth and naming the child. Ideally, discussion with the family should be conducted by one professional. The team has also the responsibility to educate other health staff in the appropriate initial management of affected newborn infant, or child and their families.
A complete detailed history is essential for an appropriate diagnosis, which include unexplained deaths, fertility, similar genital ambiguity in the family, hirsutism, amenorrhea, maternal exposure to exogenous medications, containing androgens, such as synthetic progesterone preparation and consanguinity. Also, the possibility of placental aromataze deficiency, maternal androgen secreting hormone, and a history of maternal virilization during pregnancy. Patient should be examined, in the presence of parents, where the physical findings should be demonstrated to them. In a warm and quit place, while the baby in the frog leg position, a general inspection is performed focusing on the genitalia, any other associated congenital anomalies, signs of dehydration and hyperpigmentation. Then palpate for the gonads within the inguinal canal, scrotum, and labioscrotal folds. The gonads are sometime not palpable. A palpable gonad is most likely testis or rarely ovotestis because a streak of gonads and ovaries do not usually descend. Measure the abnormal phallic width and stretched penile length, as well as the examination for fused labia or biffed scrotum and determine the position of the urethral opening and other orifices in the perineum.
The Prader classification Table 3 provides a useful guide for the severity of virilization. The finding ranges from mild clitoromegaly, Prader I, to complete virilization with urethral opening up at the tip of the enlarged phallus like structure, Prader 5. The patient can be classified, therefore, into the the palpable and non-palpable gonads. The following represents the diagnostic algorithm for patients with ambiguous genitalia in which the gonads are either palpable or not (Figure 4). Although, diagnostic algorithms do exist, yet with the spectrum of findings, no single evaluation protocol can be recommended in all circumstances. Establishing the genetic sex (karyotype) should be the first step, coupled with determining the anatomical status of the internal organs, by utilizing the various radiological tools (Ultrasound (US), Fluoroscopy-Genitography and Magnetic Resonance Imaging (MRI) (Figures 5-9). Further specific hormonal investigations (serum electrolytes, glucose, 17 – hydroxy progesterone (17-OHP), testosterone, gonadotropins and anti- Mullerian hormone). and therapeutic trials need to be undertaken to specify the cause and, hence, the appropriate therapy. Decision making algorithms based on the karyotype, and other hormonal investigation are available as a guideline [7,15-22]. Possible diagnosis and further investigations depend on the results of the initial investigations. The hGG, ACTH and GnRH stimulation tests to assess testicular and related steroid biosynthesis pathway, can be perfound.

Figure 4: Diagnostic Algorithm for patients with ambiguous genitalia, based on gonadal palpation.

Figure 5: Ultrasound pelvis demonstrates fluid filed upper vagina (white arrow). Ovaries were not visualized on this scan. The urinary bladder (white star).

Figure 6: Ultrasound pelvis demonstrates presence of the uterus (white arrow). No ovaries were visualized. The urinary bladder (white star).

Figure 7: Ultrasound pelvis (A) and scrotum (B) and (C) demonstrate absence of uterus and ovaries with presence of both testes in the scrotum (white arrows) with mild hydrocele on left side (white star). The urinary bladder (white star) and rectum (black star).

Figure 8: Fluoroscopy genitogram demonstrates normal appearing urinary bladder (black star) and urethra (white arrow) with dilated upper vagina (black arrowhead) with communication with lower urethra (black arrow).

Figure 9: (A and B) MRI pelvis T2 weighted image sagittal (A) and axial (B) demonstrates presence of a dilated upper vagina (black star) and the uterus (white arrow) with two separate cavities (white arrow heads) (bicornute uterus). No ovaries or testes were visualized.
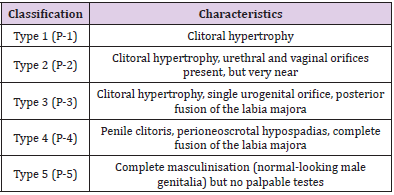
Table 3: Degree of virilisation of the external genitalia according to Prader’s classification.
Some trial of testosterone injections could differentiate between partial and complete androgen insensitivity. Gene analysis. Studies can be performed in specialized laboratories. Laparotomy or laparoscopy with gonadal biopsies are needed at times. Preoperative endoscopy and laparoscopy can provide useful anatomical details that can help in the diagnosis and surgical planning. The presence of a cervix and a uterus exclude 46, XY DSD, except for persistent Müllerian duct syndrome and dysgenetic gonads. Preoperative endoscopy is considered an integral part of genitoplasty, as it facilitates the insertion of a Fogarty balloon catheter. A biopsy as well as laparoscopy is not required when the diagnosis is clearly established biochemically or by gene studies, as the histology can be confidently predicted. It is only required when an ovotestis or dysgenetic gonad is suspected, to determine the definitive diagnosis (Figure 10) [2,24]. The assignment depends on many factors, including the genes involved. Despite the significant progress made over recent years in understanding the genetic basis of human sexual development, a specific molecular diagnosis is identified in about 20% of cases of DSD. Most virilised 46, XX infants will have CAH. By contrast, only half of 46, XY children with DSD will be given a definitive diagnosis. Nevertheless, the molecular diagnosis alone cannot dictate the gender assignment. There are several other factors that cannot be ignored, including genital appearance, prenatal androgen exposure, surgical options, the need for lifelong hormone therapy, fertility potential, family wishes, and social circumstances.

Figure 10: A medical photograph of a patient with ambiguous genitalia (A) the karyotype showed 45X/46XY a mar SRY positive. Note, normal vagina and urethra, with marked clitromegally, magnetic resonance imaging (MRI) of pelvis (B) showing hypoplastic uterus and gonadal dysgenesis. Pelvic laparscopy showed a small uterus, well developed right gonad (ovary), and left streak of gonads (testes) (C).
Disorders that are preferably raised as males are:
1. XY karyotype, relative virilisation, partial androgen insensitivity (PAIS), 5α-reductase or 17β-hydroxysteroid dehydrogenase deficiency.
a. At puberty, two-thirds of those reared as females virilise and live as males.
b. Fertility is possible in 5α-reductase deficiency.
c. Dissatisfaction is reported by 25% of patients with PAIS regardless of sex of rearing.
1. Micropenis and 46, XY cloacal exstrophy.
2. 46, XX CAH in older patients who have normal male external genitalia but with a delayed diagnosis. By contrast, evidence supports the current recommendation to raise markedly virilised 46, XX newborns with CAH as female.
Disorders that are preferably raised as females are:
a. 46, XY individuals with PAIS or genital gonadal dysgenesis (GD) with poorly virilised genitalia.
b. 46, XX individuals with CAH with milder degrees of virilisation.
c. All patients with 46, XY CAIS with no ambiguous genitalia who were assigned as female in infancy. All such patients later identify themselves as females.
Although the diagnosis of the various etiological causes of disorders pf sex development (DSD) can be delineated through chromosomal analysis, the radiological imaging, and various hormonal studies, the issue of sex assignment in the newborn with ambiguous genitalia is in a state of controversy. The conventional way to the approach of sex assignment depends on so many factors of which; the potential for sexual activity, fertility, especially in 46XX female, the feasibility of constructive surgery, and cosmetic appearance of the reconstructed genitalia based on the recognition that the external female genitalia are generally easier to construct than male. Patients with 46 XX CAH should be raised as females, because 90% of them have a female gender identify, the potential for normal sexual function, and fertility in 90%. However, they are not without long term psychosocial adjustment problems. In contract, management of patients with 46 XY DSD are so complex and need better understanding. Patients with XY karyotype and steroid 5-α-reductase, and 17-β-hydroxysteroid dehydrogenase deficiency are usually raised as males. This reflect the potentiality for virilization at puberty and the possibility for fertility, while that for patients with 46 XY and complete androgen insensitivity syndrome (CAIS) or complete gonadal dysgenesis are to be raised as females as they have usually phenotype females and require minimal surgery.
Management
Managing a patient with sexual ambiguity is so complex and challenging and depends on the sex assigned. Hormonal replacement therapy and surgery, all of which are crucial and considered as a part of the care.
Hormone Replacement Therapy
Hormonal therapy forms an important part of the treatment of disorders of sex development (DSD). For some conditions, such as, congenital adrenal hyperplasia (CAH), which forms the majority, hormonal replacement therapy is lifesaving as both hormones necessary for survival (cortisol and aldosterone). For patients with one of the many causes of hypogonadism sex hormone (testosterone) replacement therapy may be needed at puberty to stimulate sexual development. It has equally important and highly beneficial effects on bone minerals density. Dexamethase therapy is also used to prevent the development of ambiguous genitalia in female fetus with 21-hydroxylase deficiency CAH. As a matter of fact, that these hormones does nothing to treat or cure the underlying disease. For patients who were raised as females sex estrogen therapy should be prescribed, during puberty, to stimulates the sexual characteristics [25,26].
Surgery
Once an appropriate sex assignment has been made, the next critical step is the performance, if needed, of reconstructive surgery, such as feminizing genitoplasty in those rented a female sex. The timing is so crucial and important. While most surgeons prefer it to be done early in infancy, as surgery will improve the child’s maternal relationship, and relieve some of the parental distress. Other surgeons prefer to do it at puberty, Vaginal dilatation should be avoided before puberty. Children who have early reconstructive surgery should have revision of the vagina at adolescence. The testes in patients with complete adrenal insensitivity syndrome (CAIS), and other patients who were raised as females should be removed to avoid malignancy in adulthood. The time is debatable, while some recommend its removal at the time of diagnosis to avoid the adverse effects of testosterone on the neurons. Others suggest to defers that at the time of puberty to allow estrogen formation, needed for feminization so as for the streak of gonads. In individuals with 46XY karyotype, who were raised as males such as partial androgen insensitivity syndrome (PAIS) and steroid 5-α-reductaseenzyme- 2-deficiency, the management is so complex. They have shown a strong tendency to virilize at puberty. Also, it is important to understand that it is more difficult and complex to reconstruct a penis. This is usually start with releasing the chordee and constructing a urethra. Finally, a laproseay is essential to diagnose and treat some forms of disorders of sex development (DSD).
The patients with urogenital sinus complicated by urinary stasis and/ or infection may require an early intervention. The ease with which vaginoplasty can be performed at any age depends on the length of the common urogenital sinus. The essential elements of feminising genitoplasty include vaginoplasty and clitoral reduction; recession, i.e, clitoral shaft resection with preservation of the glands and its neurovascular bundles. Vaginal lengthening is important for satisfactory intercourse, various methods of selfdilation of total vaginal replacement is required, sigmoid colon or ileum are probably the best options, but both skin and amnion graft can be used [27-36].
Acknowledgement
The authors would like to thank Mr. Ibrahim N.A ALJurayyan, medical student, for his kind assistance in preparing and typing this manuscript.
Conflict Of Interest
The authors have no conflicts of interest to declare.
Funding
None.
References
- Lee PA (2004) perspective on the approach to the intersex child born with genital ambiguity. J Pediatr Endocrinol Metab 17(2): 133-140.
- Aaronson IA Aaronson AJ (2010) How should we classify intersex disorders?J Pediatr Urol 6(5): 443-446.
- Hiort O, Birnbaum W, Marshall L, Wünsch L, Werner R, et al. (2014) Management of disorders of sex development. Nat Rev Endocrinol 10: 520-529.
- Houk CP, Lee PA (2012) Update on disorders of sex development. Current Opinion in Endocrinology, Diabetes and Obesity 19(1): 28-32.
- Moshiri M, Chapman T, Fechner PY, Dubinsky TJ, Shnorhavorian M, et al. (2012) Evaluation and management of disorders of sex development: multidisciplinary approach to a complex diagnosis. Radiographics 32(6): 1599-1618.
- Chavhan GB, Parra DA, Oudjhane K, Miller SF, Babyn PS, et al. (2008) Imaging of ambiguous genitalia: classification and diagnostic approach. Radiographics 28(7): 1891-904.
- AlJurayyan N (2013) Imaging of disorder of sex development. Annals of Saudi Medicine 33(4): 363-367.
- Ahmed SF, Achermann JC, Arlt W, Balen A, Conway G, et al. (2016) Society for Endocrinology UK guidance on the initial evaluation of an infant or an adolescent with a suspected disorder of sex development (Revised 2015). Clin Endocrinol (Oxf) 84(5): 771-788.
- Wherrett DK (2015) Approach to the Infant with a Suspected Disorder of Sex Development. Pediatr Clin North Am 62(4): 983-999.
- Cools M, Nordenström A, Robeva R, Hall J, Westerveld P, et al. (2018) Caring for individuals with a difference of sex development (DSD): a Consensus Statement. Nat Rev Endocrinol 14(7): 415-429.
- Guerra-Junior G, Andrade KC, Barcelos IHK, Maciel-Guerra AT (2018) Imaging Techniques in the Diagnostic Journey of Disorders of Sex Development. Sexual Development 12(1-3): 95-99.
- Palmer BW, Wisniewski AB, Schaeffer TL, Mallappa A, Tryggestad JB, et al. (2012) model of delivering multi-disciplinary care to people with 46 XY DSD. J Pediatr Urol 8(1): 7-16.
- Morel Y, Rey R, Teinturier C, Nicolino M, Michel-Calemard L MowszowiczetI, et al. (2002) Aetiological diagnosis of male sex ambiguity: a collaborative study. Eur J Pediatr 161(1): 49-59.
- Conway GS (2014) Disorders of sex development (DSD): an overview of recent scientific advances. Psychology and sexuality 5: 28-33.
- Avni FE, Lerisson H, Lobo ML, Cartigny M, Napolitano M, et al. (2019) Plea for a standardized imaging approach to disorders of sex development in neonates: consensus proposal from European Society of Paediatric Radiology task force. Pediatr Radiol 49(9): 1240-1247.
- (2013) American Institute of Ultrasound in Medicine: AIUM practice guideline for the performance of obstetric ultrasound examinations. J Ultrasoun Med 32(6): 1083-1101.
- Ballano IG, Cornudella RS, Puertas DL, Luis JD (2015) Sonographic fetal sex determination in the first trimester: study in 2314 pregnancies and literature review. Gynecol Obstet Mex 83(4): 207-212.
- Riccabona M, Darge K, Lobo ML (2015) ESPR Uroradiology Taskforce – imaging recommendations in paediatric uroradiology, part VIII: retrograde urethrography, imaging disorder of sexual development and imaging childhood testicular torsion. Pediatr Radiol 45(13): 2023-2028.
- Al-Alwan I, Navarro O, Daneman D, Daneman A (1999) Clinical utility of adrenal ultrasonography in the diagnosis of congenital adrenal hyperplasia. J Pediatr 135(1): 71-75.
- Kanemoto K, Hayashi Y, Kojima Y, Maruyama T, Ito M, et al. (2005) Accuracy of Ultrasonography and Magnetic Resonance Imaging in the Diagnosis of non-palpable testis. Int J Urol 12(7): 668-672.
- Al Jurayyan NA, Patel PJ, al Herbish AS, Abdullah MA, Abo-Bakr AM, et al. (1995) Ambiguous genitalia: comparative role of pelvic ultrasonography and genitography. Ann Trop Paediatr 15(3): 203-207.
- Mansour SM, Hamed ST, Adel L, Kamal RM, Ahmed DM, et al. (2012) Does MRI add to ultrasound in the assessment of disorders of sex development? Eur J Radiol 81(9): 2403-2410.
- Gambino J, Caldwell B, Dietrich R, Walot I, Kangarloo H, et al. (1992) Congenital Disorders of Sexual Differentiation; MR Findings. AJR AM J Roentgenol 158(2): 363-367.
- Brian A Vanderbrink, Richard C Rink, Mark P Cain, Martin Kaefer, Kirstan K Meldrum, et al. (2010) Does preoperative genitography in congenital adrenal hyperplasia cases affect surgical approach to feminizing genitoplasty? J Urol 184(4): 1793-1798.
- Birnbaum W, Bertelloni S (2014) Sex hormone replacement in disorders of sex development. Endocr Dev 27: 149-159.
- Hewitt J, Zacharin M (2015) Hormone replacement in disorders of sex development: current thinking. Best Pract Res Clin Endocrinol Metab 29(3): 437-447.
- Mouriquand PDE, Mure PY (2004) Current concepts in hypospadiology. BJU Int 93: 26-34.
- Eroglu ETekant G, Gundogdu G, Emir H, Ercan O, Soyletet Y, et al. (2004) Feminizing surgical management of intersex patients. Pediatr Surg Int 20(7): 543-547.
- Burgu B, Duffy PG, Cuckow P, RansleyP, Wilcox D, et al. (2007) Long-term outcome of vaginal reconstruction: comparing techniques and timing. J Pediatr Uro 3(4): 316-320.
- Callens N, Hoebeke P (2014) Phalloplasty: a panacea for 46, XY disorder of sex development conditions with penile deficiency? Endocr Dev 27: 222-233.
- Michala L, Liao L-M, Wood D, Conway GS, Creighton SM, et al. (2014) Practice changes in childhood surgery for ambiguous genitalia? J Pediatr Urol 10(5): 934-939.
- Wolffenbuttel KP, Crouch NS (2014) Timing of feminising surgery in disorders of sex development. Endocr Dev 27: 210-221.
- Mouriquand P, Caldamone A, Malone P, Frank JD, Hoebeke P, et al. (2014) The ESPU/SPU standpoint on the surgical management of Disorders of Sex Development (DSD). J Pediatr Urol 10(1): 8-10.
- Kutney K, Konczal L, Kaminski B, Uli N (2016) Challenges in the diagnosis and management of disorders of sex development, Birth Defects Research 108(4): 293-308.
- Lee P, Schober J, Nordenstrom A, Hoebeke P, Houk C, et al. (2012) Review of recent outcome data of disorders of sex development (DSD): emphasis on surgical and sexual outcomes. J Pediatr Urol 8(6): 611-615.
- van der Zwan YG, Janssen EHCC, Callens N, Wolffenbuttel KP, Cohen-Kettenis PT, et al. (2013) Severity of virilization is associated with cosmetic appearance and sexual function in women with congenital adrenal hyperplasia: a cross-sectional study. J Sex Med 10(3): 866-875.
