 Review Article
Review ArticleAbstract
Unchecked-activation of immune responses leads to excessive expression of NF-kB, which further leads to multi organ failure at the site of viral infection and systemic level. Several studies showed that elevated expression of NF-kB related genes further amplifies inflammatory soluble cytokine/chemokines worsening the COVID infection. Although the prospective of counteracting cytokines flood is compelling, a significant limitation depends on understanding the immune signaling pathways triggered by COVID. Thus, finding more clues about the immune system’s role in COVID-19 management may prevent the transition from mild to severe stages.
Keywords: COVID-19; TLR4-MD2; Glucocorticoids; IL-6, TNF-α; Cytokine; Chemokines
Abbreviations: SARS-CoV-2: Severe Acute Respiratory Syndrome Coronavirus 2; MERS: Middle East Respiratory Syndrome; ARDS: Acute Respiratory Distress Syndrome; ACE2: Angiotensin Converting Enzyme II; CRS: Cytokine Release Syndrome; TLR4: Toll- Like Receptor 4; PAMPs: Pathogen-Associated Molecular Patterns; DAMPs: Damage- Associated Molecular Patterns; GCs: Glucocorticoids; AP1: Activator Protein 1; Jak-STAT: Janus Kinase-Signal Transducer and Activator Of Transcription; PBMCs: Peripheral Blood Mononuclear Cells; DDKs: Diphenyldihaloketones
Introduction
In the last two decades, severe acute respiratory syndrome (SARS) and the Middle East respiratory syndrome (MERS) caused severe respiratory diseases in endemic areas. However, the most recent coronavirus epidemic dwarfs SARS and MERS in its social, medical, and economic impact. Discovered in December 2019 in Wuhan, Hubei province, China, this highly contagious respiratory syndrome was given an acronym of COVID-19 (coronavirus disease 2019) to stands for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Essentially, COVID-19 is a respiratory illness with the lung as the target organ. Severe and irreparable structural and functional damage to the lung is common, which phenotypically results in acute respiratory distress syndrome (ARDS). What sets COVID-19 apart from SARS-CoV and MERS are its contagious nature, ability to survive on inanimate objects for prolonged times, rapid disease progression, and non-responsiveness to conventional anti-viral therapies. Till November 2020, COVID-19 has caused 62,363,527 active cases and 1,456,687 confirmed deaths in 220 countries [1]. These numbers are projected to rise unless an effective vaccine is globally and strategically implemented promptly. Even if the vaccine is made universally available, therapeutic alternatives to treat existing infection and its clinical sequel will be the key to win the ongoing fight against COVID-19.
About 30% of patients with COVID-19 infection in ICU would develop ARDS with characteristic severe lung edema, dyspnea, hypoxemia, and about 65% of these patients with ARDS died. Typical treatment of ARDS is based on pharmacologic modulation of the innate immune system, and it is administered to understand that ARDS is a syndrome of hyper-inflammation. Although inflammation is a protective innate immune response against viral insult, exaggerated inflammation or persistent inflammatory feed-forward loop is highly damaging to the lung and contributes to its failure [2]. However, ARDS is a heterogeneous manifestation because different patients can respond differently to the same treatment; thus, efforts have been made to segregate patients into subgroups depending on clinical outcomes [3]. Moreover, COVID-19 patients with ARDS eventually develop multi-organ failure. Unfortunately, there is no definite and clinically validated drug for ARDS capable of reducing either short-term distress or longterm mortality in COVID-19 patients. SARS-CoV-2 uses angiotensin converting enzyme II (ACE2) as its entry receptor with the help of transmembrane serine protease 2 (TMPRSS2), suggesting ACE2, and TMPRSS2 as potential therapeutic targets for COVID-19 [4,5]. These targets may serve at the initial phase of infection. Still, they may not work during the later phase of the disease when coercive chronic inflammation induced in the disease progression phase and ARDS become the main pathologic features from a clinical perspective (Figure 1).
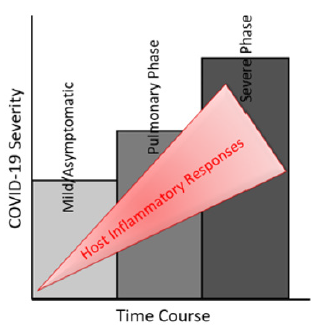
Figure 1: A tri-phasic model for understanding COVID-19 progression.
Putative Anti-Inflammatory Targets for ARDS Associated with COVID-19
At the causal level, ARDS is viewed as a cytokine release syndrome (CRS). The central signaling mechanism involved in creating cytokine storms is the hyper-activated nuclear factorkappa B (NFκB) pathway [6]. Therefore, attention has been focused on restraining or attenuating the magnitude and duration of NFκB-mediated signaling. In a recent intriguing report, Sohn, et al. implicated S100A9, an alarmin and Toll-like receptor 4 (TLR4) ligand for the activation of NFκB pathway in COVID-19 patients [7]. In general, TLRs recognize pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) to activate innate immune response; TLR3, TLR7, TLR8, and TLR9 also recognize viral components (Figure 2) [8-10].
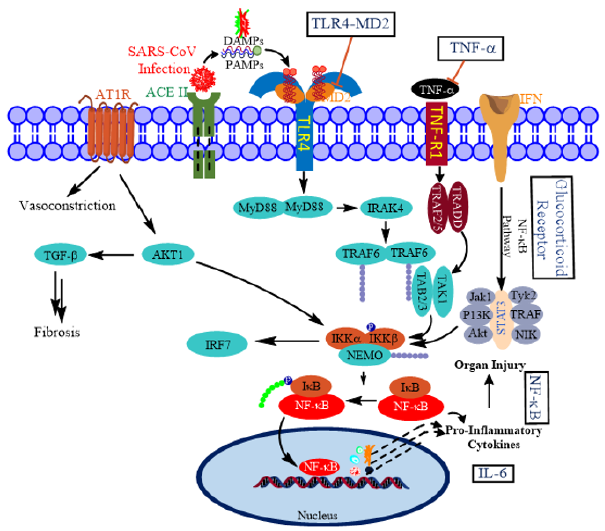
Figure 2: Putative molecular targets for restraining cytokine storm in COVID-19. SARS-CoV-2 uses angiotensin converting enzyme II (ACE2) as a cell entry receptor. The infection results in cytokine release syndrome through various pathways pivoted by NFκB. NFκB pathway is activated by the interaction of PAMPS and DAMPs with their cell-surface receptors. Boxed titles are possible therapeutic targets for COVID-19. NFkB and JAK-STAT signaling pathways are crucial for a proper viral response.
Glucocorticoids (GCs) Restrain Inflammatory Response in COVID-19 Patients
Glucocorticoids are potent anti-inflammatory and immunosuppressive agents. Their use has been recommended for severe cases of COVID-19 [11], and positive outcomes have been reported in a few clinical studies. For instance, findings from a controlled clinical trial involving patients with COVID-19 pneumonia indicated that the use of methylprednisolone showed a beneficial effect in severe COVID-19 pneumonia [12]. Similarly, in another randomized clinical study in COVID-19 patients with moderate-to-severe ARDS, dexamethasone treatment increased the survival rate in hospitalized patients supported by supplemental oxygen or mechanical ventilation[13]. Upon binding of GCs, the glucocorticoid receptor (GR) undergoes phosphorylation and nuclear translocation, where it binds to GC‐response elements (GREs) and regulates transcription. Among the three GREs (simple, composite, and tethering), recruitment of GR to the tethering site represses activities of pro-inflammatory transcription factors activator protein 1 (AP1) and NFκB.
Whereas inhibition of AP-1 and NFκB is an important mechanism of GC action, it only partially explains the immunosuppressive and potent anti-inflammatory activity of GCs. Glucocorticoids modulate cytokine activity via Janus kinase-signal transducer and activator of transcription (Jak-STAT) pathway [14,15]. The Jak-STAT pathway is activated when cytokines bind their cell surface receptor, which leads to the phosphorylation-dependent activation of STAT proteins [14]. Multiple ligands can activate individual STAT proteins, and specific cytokines can also preferentially activate particular STATs. For example, interleukin‐6 (IL‐6) preferentially activates STAT3, whereas IFNγ preferentially activates STAT1. It’s been reported that GCs and IL-6-STAT3 axis synergizes with each other. Bagca and Avci recently reviewed the potential of inhibition of the Jak- STAT pathway by ruxolitinib to treat COVID-19 [16]. On the other hand, Meyer, et al. described a combined use of GCs and ruxolitinib for fighting cytokine storm [17], which could also be applicable in COVID-19 [18].
Interleukin (IL)-6 as a Target for Dampening the Hyperactive Immune System in COVID-19
Emerging clinical trial data suggest that immunomodulatory drugs can reduce the cytokine storm. IL-6 is a pro-inflammatory cytokine produced by several cell types, and its production is elevated COVID-19 affected lung tissue [19,20]. It has been rationalized that inhibition of IL-6 or its effects may alter the course of COVID-19. The impact of IL-6 could be inhibited either by antibodies against IL-6 or its receptor (IL-6R). The Food and Drug Administration-approved monoclonal antibodies against IL-6R include sarilumab and tocilizumab, whereas siltuximab is a monoclonal antibody against IL-6. The complete list of such antibodies at various stages of clinical trials is given in Table 1.
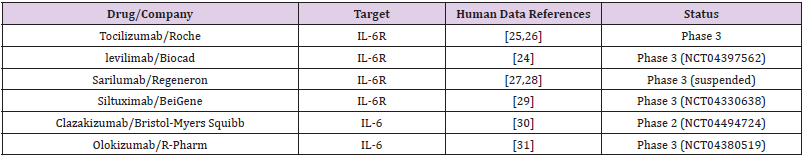
Table 1:Monoclonal antibodies against IL-6 or IL-6R under clinical trials as of 30 October, 2020.
A recent retrospective study found that tocilizumab decreased C-reactive protein in most of the patients [21]. Another retrospective study demonstrated that tocilizumab effectively controlled the severity of COVID-19 symptoms such as fever, respiratory illness [22]. Levilimab is another antibody against IL-6R, which is currently in clinical testing against COVID-19 [23]. The Russian Federation registered levilimab (marked under the name Ilsira) to control cytokine storm associated with COVID-19 [24]. Despite these initial successes, the composite data from various trials have not been promising so far, which led the COVID-19 Treatment Guidelines Panel to recommend against the use of anti-IL-6 receptor monoclonal antibodies or anti-IL-6 monoclonal antibody for the treatment of COVID-19, except in a clinical trial (Table 1).
Anti-TNF-α Therapies for COVID-19 Treatment
TNF-α is essential in nearly all acute inflammatory reactions, acting as an amplifier of inflammation. In COVID-19 patients, serum level of TNF-α at the time of hospitalization was a strong predictor of patient survival (Del Valle 2020 Nature Med 26: 1636). Interestingly, TNF blockade leads to downregulation of other pro-inflammatory mediators, including IL-1, IL-6, and granulocyte-macrophage colony stimulating factor, suggesting that anti-TNF-α therapies might be an attractive approach to restrain cytokines in COVID-19 patients [32,33]. Clinical evidence from other inflammatory diseases also suggests that anti-TNF therapies could benefit COVID-19 management [34,35]. However, very few investigations are examining anti-TNF therapies for COVID-19 treatment (Table 2).
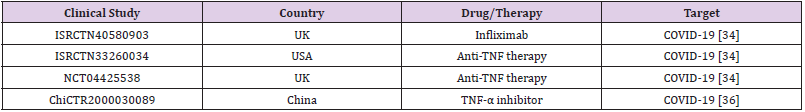
Table 2:Anti TNF-α in clinical trials against COVID-19.
Blocking Upstream Regulators of NFκB Pathway
A recent study from Korea showed that toll-like receptor (TLR) 4-mediated inflammatory signaling pathways, which mimic pathogenesis of bacterial sepsis, were highly upregulated in peripheral blood mononuclear cells (PBMCs) from COVID-19 patients, compared with healthy controls [7]. Among the most highly increased inflammatory mediators, S100A9, a TLR4 ligand, was found as a biomarker, suggesting TLR4 signaling may contribute to COVID-19 inflammation. Curcumin-like chalcones were reported to modulate MD2-TLR4 binding nullified TLR4 activation and sequentially inhibited NF-kB activation [37,38], suggesting these MD2 modulators can be used therapeutic purposes through targeting TLR4-mediated inflammation. Several small molecules reported as NF-κB inhibitors, such as diphenyldihaloketones (DDKs), namely EF24 and CLEFMA may be helpful in controlling “cytokine storm”. EF24 shown to affect several functions of DCs, besides reduces NF-κB activation and suppresses pro-inflammatory cytokine secretion [39-42].
Authors Contribution
B Chidipi and V Eada are designed this concept and wrote this paper.
Consent for Publication
Not applicable.
Acknowledgment
None.
Funding Information
The American Heart Association Postdoctoral grant (19POST34450203) supported to Bojjibabu Chidipi.
Conflict of Interests
The authors declare that they have no conflict of interests.
References
- WHO (2020) Coronavirus Disease (COVID-19) Dashboard. World Health Organization.
- Garlanda C, Charles A Dinarello, A Mantovani (2013) The Interleukin-1 Family: Back to the Future. Immunity 39(6): 1003-1018.
- Fan E, D Brodie, AS Slutsky (2018) Acute Respiratory Distress Syndrome: Advances in Diagnosis and Treatment. Jama 319(7): 698-710.
- Kuba K, Imai Y, Rao S, Gao H, Guo F, et al. (2005) A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nature Medicine 11(8): 875-879.
- Hoffmann M, Kleine Weber H, Schroeder S, Krüger N, Herrler T, et al. (2020) SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 181(2): 271-280.e8.
- Pahl HL (1999) Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 18(49): 6853-6866.
- Sohn KM, Lee SG, Kim HJ, Cheon S, Jeong H, et al. (2020) COVID-19 Patients Upregulate Toll-like Receptor 4-mediated Inflammatory Signaling That Mimics Bacterial Sepsis. J Korean Med Sci 35(38): e343.
- Kawai T, S Akira (2010) The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nature Immunology 11(5): 373-384.
- Arpaia N, GM Barton (2011) Toll-like receptors: key players in antiviral immunity. Current Opinion in Virology 1(6): 447-454.
- Doyle SL, LA O'Neill (2006) Toll-like receptors: from the discovery of NFkappaB to new insights into transcriptional regulations in innate immunity. Biochem Pharmacol 72(9): 1102-1113.
- Zhao JP, Hu Y, Du RH, Chen ZS, Jin Y, et al. (2020) Expert consensus on the use of corticosteroid in patients with 2019-nCoV pneumonia]. Zhonghua Jie He Hu Xi Za Zhi 43(3): 183-184.
- Corral L, Bahamonde A, Arnaiz Revillas F, Gómez Barquero J, Abadía Otero J, et al. (2020) GLUCOCOVID: A controlled trial of methylprednisolone in adults hospitalized with COVID-19 pneumonia. MedRxiv, p. 1-24.
- The RECOVERY Collaborative Group (2021) Dexamethasone in Hospitalized Patients with Covid-19. New England Journal of Medicine 384: 693-704.
- Rogatsky I, LB Ivashkiv (2006) Glucocorticoid modulation of cytokine signaling. Tissue Antigens 68(1): 1-12.
- Bianchi M, C Meng, LB Ivashkiv (2000) Inhibition of IL-2-induced Jak-STAT signaling by glucocorticoids. Proceedings of the National Academy of Sciences 97(17): 9573-9578.
- Quiros Roldan E, G Biasiotto, I Zanella (2020) Letter to the Editor on “Bonafè M, Prattichizzo F, Giuliani A, Storci G, Sabbatinelli J, Olivieri F. Inflamm-aging: Why older men are the most susceptible to SARS-CoV-2 complicated outcomes. Cytokine Growth Factor Rev”. Cytokine & Growth Factor Reviews 54: 1-2.
- Meyer LK, Verbist KC, Albeituni S, Scull BP, Bassett RC, et al. (2020) JAK/STAT pathway inhibition sensitizes CD8 T cells to dexamethasone-induced apoptosis in hyperinflammation. Blood 136(6): 657-668.
- Cron RQ (2020) One-two punch of cytokine storm syndrome. Blood 136(6): 645-646.
- Zhou F, Yu T, Du R, Fan G, Liu Y, et al. (2020) Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. The Lancet, 2020. 395(10229): 1054-1062.
- Wang Z, Yang B, Li Q, Wen L, Zhang R (2020) Clinical Features of 69 Cases With Coronavirus Disease 2019 in Wuhan, China. Clin Infect Dis 71(15): 769-777.
- Hassoun A, Thottacherry ED, Muklewicz J, Aziz QUA, Edwards J (2020) Utilizing tocilizumab for the treatment of cytokine release syndrome in COVID-19. J Clin Virol 128: 104443.
- Xu X, Han M, Li T, Sun W, Wang D, et al. (2020) Effective treatment of severe COVID-19 patients with tocilizumab. Proceedings of the National Academy of Sciences 117(20): 10970-10975.
- Twomey JD, Luo S, Dean AQ, Bozza WP, Nalli A, et al. (2020) COVID-19 update: The race to therapeutic development. Drug Resistance Updates 53: 100733.
- Harrison C (2020) Focus shifts to antibody cocktails for COVID-19 cytokine storm. Nature Biotechnology 38(8): 905-908.
- Luo P, Liu Y, Qiu L, Liu X, Liu D, et al. (2020) Tocilizumab treatment in COVID-19: A single center experience. Journal of Medical Virology 92(7): 814-818.
- Guaraldi G, Meschiari M, Cozzi Lepri A, Milic J, Tonelli R, et al. (2020) Tocilizumab in patients with severe COVID-19: a retrospective cohort study. The Lancet Rheumatology 2(8): e474-e484.
- Benucci M, Giannasi G, Cecchini P, Gobbi FL, Damiani A, et al. (2020) COVID-19 pneumonia treated with Sarilumab: A clinical series of eight patients. Journal of Medical Virology 92(11): 2368-2370.
- Della Torre E, Campochiaro C, Cavalli G, De Luca G, Napolitano A, et al. (2020) Interleukin-6 blockade with sarilumab in severe COVID-19 pneumonia with systemic hyperinflammation: an open-label cohort study. Annals of the Rheumatic Diseases 79(10): 1277-1285.
- Gritti G, Raimondi F, Ripamonti D, Riva I, Landi F, et al. (2020) IL-6 signalling pathway inactivation with siltuximab in patients with COVID-19 respiratory failure: an observational cohort study. medRxiv, p. 1-31.
- Vaidya G, Czer LSC, Kobashigawa J, Kittleson M, Patel J, et al. (2020) Successful Treatment of Severe COVID-19 Pneumonia With Clazakizumab in a Heart Transplant Recipient: A Case Report. Transplantation Proceedings 52(9): 2711-2714.
- Genovese MC, Fleischmann R, Furst D, Janssen N, Carter J, et al. (2014) Efficacy and safety of olokizumab in patients with rheumatoid arthritis with an inadequate response to TNF inhibitor therapy: outcomes of a randomised Phase IIb study. Annals of the Rheumatic Diseases 73(9): 1607-1615.
- Duret PM, Sebbag E, Mallick A, Gravier S, Spielmann L, et al. (2020) Recovery from COVID-19 in a patient with spondyloarthritis treated with TNF-alpha inhibitor etanercept. Annals of the Rheumatic Diseases 79(9): 1251-1252.
- Feldmann M, Maini RN, Woody JN, Holgate ST, Winter G, et al. (2020) Trials of anti-tumour necrosis factor therapy for COVID-19 are urgently needed. The Lancet 395(10234): 1407-1409.
- Robinson PC, Richards D, Tanner HL, Feldmann M (2020) Accumulating evidence suggests anti-TNF therapy needs to be given trial priority in COVID-19 treatment. The Lancet Rheumatology 2(11): e653-e655.
- Stallmach A, Kortgen A, Gonnert F, Coldewey SM, Reuken P, et al. (2020) Infliximab against severe COVID-19-induced cytokine storm syndrome with organ failure—a cautionary case series. Critical Care 24(1): 444.
- Duret PM, Sebbag E, Mallick A, Gravier S, Spielmann L, et al. (2020) Recovery from COVID-19 in a patient with spondyloarthritis treated with TNF-alpha inhibitor etanercept. Annals of the Rheumatic Diseases 79(9): 1251-1252.
- Roh E, Lee HS, Kwak JA, Hong JT, Nam SY, et al. (2011) MD-2 as the target of nonlipid chalcone in the inhibition of endotoxin LPS-induced TLR4 activity. J Infect Dis 203(7): 1012-1020.
- Venkateswararao E, Sharma VK, Lee KC, Sharma N, Park SH, et al. (2012) A SAR study on a series of synthetic lipophilic chalcones as Inhibitor of transcription factor NF-κB. European Journal of Medicinal Chemistry 54: 379-386.
- Vilekar P, Awasthi S, Natarajan A, Anant S, Awasthi V (2012) EF24 suppresses maturation and inflammatory response in dendritic cells. International Immunology 24(7): 455-464.
- Vilekar P, Rao G, Awasthi S, Awasthi V (2015) Diphenyldifluoroketone EF24 Suppresses Pro-inflammatory Interleukin-1 receptor 1 and Toll-like Receptor 4 in lipopolysaccharide-stimulated dendritic cells. Journal of Inflammation 12(1): 55.
- Sahoo K, Dozmorov MG, Anant S, Awasthi V (2012) The curcuminoid CLEFMA selectively induces cell death in H441 lung adenocarcinoma cells via oxidative stress. Investigational New Drugs 30(2): 558-567.
- Lagisetty P, Vilekar P, Sahoo K, Anant S, Awasthi V (2010) CLEFMA-an anti-proliferative curcuminoid from structure-activity relationship studies on 3,5-bis(benzylidene)-4-piperidones. Bioorg Med Chem 18(16): 6109-6120.
