 Research Article
Research ArticleAbstract
During extended myocardial ischemia combinations of hypoxia and acidosis drive cardiac cell loss by apoptosis and necrosis that contribute importantly to myocardial infarction (MI). Here we investigated roles for miR-126 in stress kinase activation, induction of inflammatory cytokines and apoptosis caused by exposure of cultured cardiac myocytes to hypoxia/acidosis or acidosis alone. Key apoptotic pathway intermediates and inflammation markers IL-6, IL-8, and tumor necrosis factor (TNF) were quantified by western blot analyses, while miR-126 expression was tracked by quantitative polymerase chain reaction (qPCR). Cause and effect of miR-126 was evidenced by knockdown (KD) with a miR-126-selective antimiR. Obligatory roles for acidosis in promoting apoptosis confirmed our previous reports that hypoxia alone does not confer a lethal signal. Acidosis with or without hypoxia increased apoptosis that was paralleled by elevated miR-126, increased phosphorylation of p38-MAPK and JNK, enhanced expression of IL-6, IL-8, and TNF, and downregulation of Bcl-2. All effects were prevented by prior transfection of myocytes with antimiR-126. The results support significant roles for miR-126 in regulating cardiac myocyte survival pathways and cell death during exposure to simulated ischemia and acidosis.
Keywords: Acidosis; miR-126; Inflammation; MAPK/JNK; Apoptosis; Myocardial Infarction
Introduction
Cardiac myocyte cell death by apoptosis accompanies heart disease of both ischemic and nonischemic origin [1-3]. It has been demonstrated in the myocardium from failing human hearts [4,5], in patients with arrhythmogenic right ventricular dysplasia [6], and in association with myocardial infarction, both within the infarcted area itself and in the surrounding viable tissue [7]. In animal models, increased apoptosis accompanies both phases of ischemia and reperfusion [8]. Our group reported that severe chronic hypoxia alone does not cause apoptosis of cardiac myocytes in culture. Rather, coincident acidosis and/or reoxygenation is required to trigger apoptosis [9-11]. Important roles of inflammatory mediators in cardiac cell death by ischemia with or without reperfusion are well established [12-14]. Proton pumps and pH regulation are known to play important roles in apoptosis signaling [15,16]. Hypoxic cardiac myocytes generate excess H+ through increased anaerobic glycolysis, net hydrolysis of ATP, and CO2 retention. Protons are extruded from the myoplasm to the interstitial space by the combined action of 3 major ion-specific membrane transporters, including the Na+/H+ exchanger, Na+/ HCO3 cotransporter, and vacuolar proton ATPase [17,18]. Increased activity of the Na+/H+ exchanger can cause Ca2+ overload because the elevated intracellular Na+ is subsequently exchanged for Ca2+ via the Na+/Ca2+ exchanger. Inhibition of Na+/H+ exchange is protective against ischemic injury, while inhibition of the vacuolar ATPase promotes apoptosis, in part by shifting the proton load toward the Na+/H+ transporter and thus increasing Ca2+ uptake, and in part by reducing the myocyte capacity to control [pH]i. Acidosis has been shown to correlate with apoptosis in a number of other systems [19-21].
MicroRNAs (miRs) are endogenous, single-stranded, noncoding RNAs that range in length from 18 to 24 nucleotides and regulate gene expression primarily by binding to the 3’ untranslated region of target genes [22,23]. The miR-126 locus generates 2 mature miRNAs, miR-126-3p and miR-126-5p [24]. Previous work linked miR-126 with vascular integrity and angiogenesis and described its downregulation, and possible biomarker role in AMI [25-28]. MiR-126 is enriched in endothelial cells (EC) and EC progenitors, and has been assigned essential roles in cardiac developmental as well as in the recovery of the heart from injury by activating survival kinases ERK1/2 and Akt and increasing proangiogenic signaling [29-31]. Roles for mirR-126 have also been described in the regulation of cardiac myocyte proliferation and inflammation, including Interleukins in non-endothelial H9c2 cells [32]. Aberrant expression of miR-126 associated with vascular pathologies including but not limited to cancers, diabetes and ocular disease has revealed roles in a range of biological processes including apoptosis, autophagy, pathological angiogenesis, cell migration and proliferation [33,34]. Roles for miR-126 in the inflammatory responses of cardiac myocytes subjected to simulated ischemia have not been fully described. We previously reported that hypoxia/acidosis but not hypoxia alone presents a powerful apoptotic stimulus for cultured cardiac myocytes [10,11,35,36]. Therefore, to further define the molecular pathway of cell death in this model we quantified miR-126 levels, markers of inflammation, survival kinases and apoptosis pathway components in cardiac myocytes subjected to acidosis with or without hypoxia and in the presence and absence of antimiR-126. The results identify miR-126 as a regulator and potential biomarker of ischemic cardiac injury.
Materials and Methods
Reagents
Antibodies were obtained from the following vendors: IL-6 (sc-130326); IL-8 (sc-32817); TNFα(sc-130349); Bcl-2 (sc-7382); p38 (sc-33688); phosphorylated (p-) p38 (sc- 166182); JNK (sc- 7345); p-JNK (sc-293136) from Santa Cruz biotechnology, GAPDH from Cell Signaling Technology. Human pre-microRNA expression constructs Anti-miR-126 was from System Bioscience LLC.
Cardiac Myocytes Culture and Exposure to Acidosis Condition
All procedures involving animals were performed in accordance with Southern Medical University guidelines for the care and use of animals. Cardiac myocytes were prepared and cultured from hearts of 1–3-day-old neonatal rat pups as previous described [10,11,35]. Briefly, after 3–5 days in culture contracting monolayers were placed in serum-free medium and exposed to treatments 72h later. Cells were cultured in Dulbecco’s Modified Eagle’s Medium, supplemented with 0.1 mg/ml G-418 and 10% fetal bovine serum in a humidified atmosphere with 5% CO2 at 37°C. Our conditions for exposure to hypoxia (0.5% O2/5% CO2) and measurement of pH are described in detail elsewhere [10,11,36].
Cell Transfection
Myocytes were transfected with anti-miR-126 plasmid and control plasmid respectively by lipofectin reagent as described by the manufacturer (Thermo Fisher Scientific). MiR-126 expression was confirmed by qPCR assay at 24h after transfection.
Apoptosis Assay
Apoptosis assays were performed using an ApopTag peroxidase in situ apoptosis detection kit (Cat# S7100, Millipore, Billerica, MA, USA), according to the manufacturer’s recommendations. Briefly, fixed cells were quenched in PBS with 3% hydrogen peroxide and incubated in equilibration buffer, working strength TdT enzyme, anti-Digoxigenin conjugate, followed by color developer peroxidase substrate and counterstain with 0.5% (w:v) methyl green. Apoptotic cells were quantified under phase contrast microscopy.
RNA Extraction and Quantitative Polymerase Chain Reaction (qPCR)
Total RNA was extracted from cell pellets using Trizol by standard procedures (Invitrogen); reverse transcription of miR- 126 expression was performed using a MultiScribe RT kit (Applied Biosystems) according to the manufacturer’s instructions. RT-PCR conditions were as follows: 3-min incubation at 95°C, followed by 40 cycles of 95°C x 15-sec, 60°C for 30-sec, and 72°C for 45-sec, followed by 5-min incubation at 4°C. Primer sequences were as follows:miR-126forward,5′-TATAAGATCTGAGGATAGGTGGGTTCC CGAGAACT-3′;reverse,5′-ATATGAATTCTCTCAGGGCTATGCCGCCT AAGTAC-3′ ; U6 forward 5’-CTCGCTTCGGCAGCACA-3’, U6 reverse 5’-AACGCTTCACGAATTTGCGT-3’.
Protein Assay
Proteins were quantified by BCA Protein Assay Kit (Pierce, Appleton, Wisconsin). and detected by standard Western blots stained with horseradish peroxidase labelled secondary antibodies.
Statistical Analysis
All experiments were repeated at least three times. Data are presented as the mean and standard deviation. Statistical comparisons were performed using ANOVA followed by paired, one-tailed t test, using InStat software for Macintosh (GraphPad Software Inc, San Diego, CA USA). Statistical difference between groups was calculated by using Student’s t-test. A p-value <0.05 was considered to be statistically significant.
Results
Hypoxia/Acidosis Augments miR-126 Expression Coincident with Increased Apoptosis
Control and starting media pH were 7.4. Progression to pH 7.2 indicated mild acidosis after 12h hypoxia/acidosis that declined to pH 6.7 after 72h (Figure 1a). MiR-126 expression was significantly increased by hypoxia/acidosis at 12h, and maximally increased by >4-fold at 72h (Figure 1b). Apoptosis assays confirmed linearly increased apoptotic cells during 72h incubations. (Figure 1c,1d). Inflammatory cytokines TNFα, IL-6, and IL-8 were quantified in lysates from cardiac myocytes subjected to identical conditions described above. As shown in (Figure 1e-1h), levels of TNFα peaked after only 12h of hypoxic/acidosis incubation whereas the levels of IL-6, and IL-8 were maximally increased after 72h coincident with the maximal induction of miR-126 (all p<0.01).
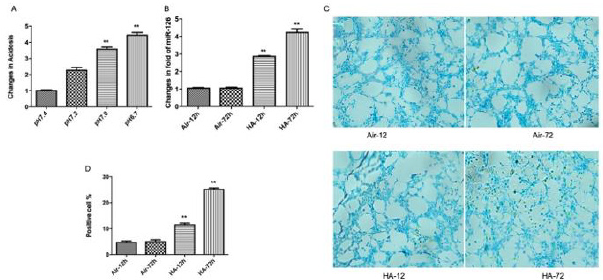
Figure 1: Hypoxia/acidosis augments miR-126 expression coincident with increased apoptosis and effect on inflammatory responses. Cultured cells were treated from acidosis-12 hours to 72 hours(A). RT-PCR showing the expression level of miR-126 (B); Cell apoptosis assay using ApopTag peroxidase in situ detection kit (C,D); Western blot revealing the expression levels of TNF-a, IL-6 and IL-8 proteins (E-H). D Data are expressed as mean+SEM. **P<0.01 compared between HA-12 h and HA-72 h.
Inhibition of Hypoxia/Acidosis-Induced Apoptosis and Inflammation by KD of miR-126
To investigate a possible causal-effect of enhanced miR-126 expression on the regulation of inflammation markers and cardiac myocyte apoptosis by hypoxia/acidosis and aerobic acidosis, we repeated the experiments of (Figures 1 & 2) in the presence of antimiR-126 to knockdown (KD) mir-126 expression in hypoxic/ acidotic and aerobic-acidosis treated myocyte cultures (Figure 2a). Under conditions of mir-126 KD, apoptotic indices were decreased, consistent with a role for miR-126 in promoting apoptosis in these models (Figure 2b, 2c). In parallel, expression levels of IL-6. IL-8, and TNFα were each significantly decreased by miR-126 KD. Therefore, elevated miR-126 expression appears to be a precondition both for induction of inflammatory factors and increased apoptosis in response to hypoxia/acidosis and aerobic acidosis (Figure 2d-2g). To investigate the timeline and possibly differential regulation of inflammation and apoptosis by miR-126 in this model, we blocked miR-126 expression at different stages after induction of hypoxia/acidosis. As shown in (Figure 3a-3d), despite quantitative differences in the responses of individual proinflammatory cytokines to incubation time and media pH, in all cases miR-126 KD conferred a greater relative suppression at the earlier stages of acidosis compared with the more severe late-stage acidosis of pH 6.7, (Figure 3e-3h). These results provide evidence for a graded effect of acidosis on inflammatory cytokine induction as well as a requirement for elevated miR-126 in the cytokine activation pathways.
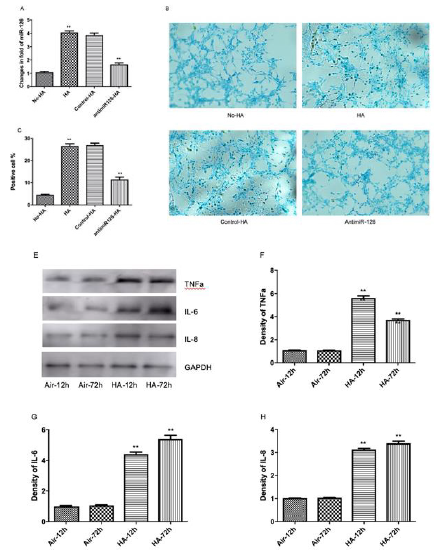
Figure 2A:

Figure 2: Inhibition of hypoxia/acidosis-induced apoptosis and inflammation by KD of miR-126. Cultured cells were treated from acidosis 72 hours with or without transfected with antimir-126 plasmids. RT-PCR showing the expression level of miR- 126 (A); Cell apoptosis assay using ApopTag peroxidase in situ detection kit (B-C); Western blot revealing the protein expression levels (D-G). Data are expressed as mean+SEM. **P<0.01 compared between No-HA and HA.
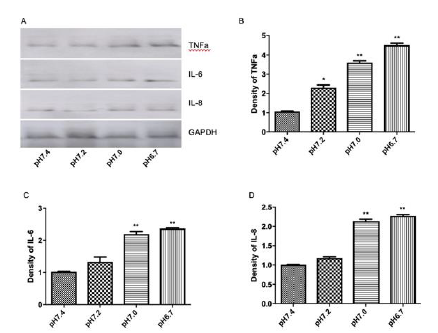
Figure 3A: The home screen of the labelling system (left) and an example of the labelling process using the system (right).
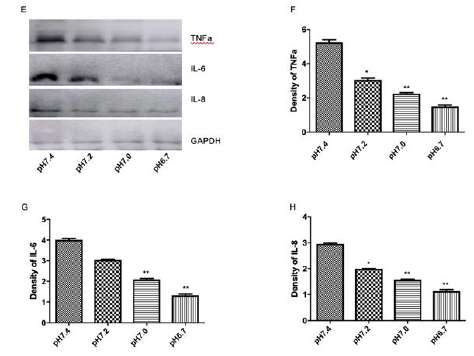
Figure 3: Roles for Bcl2 and MAPK/JNK in miR-126-mediated inflammation and apoptosis. Data are expressed as mean + SEM; Cell apoptosis assay using ApopTag peroxidase in situ detection kit (I-L);**P<0.01, *P<0.05 compared when pH=7.4, pH=7.2, pH=7.0 and pH=6.7.
Roles for Bcl2 and MAPK/JNK in miR-126-Mediated Inflammation and Apoptosis
Previous work from others and ourselves established key roles for Bcl-2, mitogen-activated protein kinase (MAPK) and c-Jun N-terminal kinase (JNK) in apoptosis pathways of cardiac myocytes exposed to redox stress, acidosis and simulated ischemia with and without reperfusion [35-39]. Regulatory roles for miR-126 have also been described in cancer cells undergoing Bcl-2-mediated apoptosis [40-42]. Therefore, we investigated a possible role for Bcl-2 in the miR-126/pH sensitive apoptotic pathway of cardiac myocytes described herein. As shown in (Figure 4a,4b), Bcl-2 levels decreased markedly in cardiac myocytes subjected to hypoxia/ acidosis or aerobic acidosis, and the effects were blocked by KD of miR-126 in parallel with reduced apoptosis. The results suggest negative regulation of anti-apoptotic Bcl-2 by miR-126 in this model that is consistent with some previous results [41,43,44].
We quantified the expression levels of p38-MAPK and JNK in hypoxic/acidotic and aerobic-acidotic cardiac myocytes in the presence and absence of miR-126 KD. As shown in Figure 4a-4d, phosphorylation of p38 and JNK increased in myocytes that were exposed to both conditions of acidosis, effects that were again blocked by KD of miR-126. The results suggest that hypoxia/ acidosis activates MAPK/JNK signaling in a manner that depends on miR-126 expression and correlates with downregulated Bcl- 2 expression. Therefore, apoptosis driven by acidosis with or without concurrent hypoxia of cardiac myocytes requires increased expression of miR-126 and MAPK/JNK, and suppression of Bcl-2.
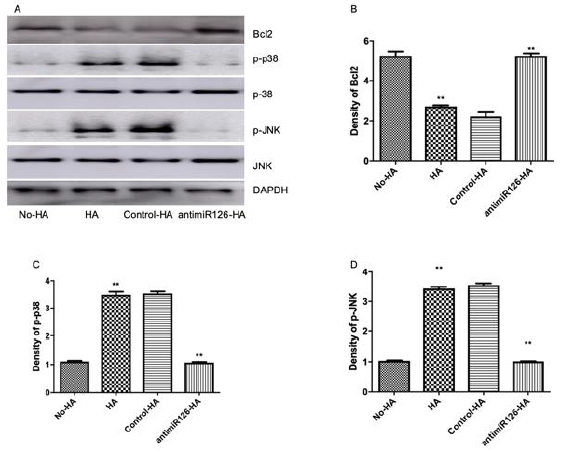
Figure 4: Regulation of Bcl2 expression by miR-126 mediated inactivation of MAPK/JNK signaling. Western blot revealing the protein expression levels (A-D); Data are expressed as mean + SEM. **P<0.01 compared between No-HA and HA.
Discussion
Apoptotic, necrotic and autophagic cell death pathways play central roles in tissue injury caused by ischemia-reperfusion during myocardial infarction (MI) and are important therapeutic targets [14,45,46]. During ischemia caused by coronary artery occlusion, affected myocardial tissues downstream of the occlusion become progressively hypoxic and acidotic [11,35,47]. In extreme cases of prolonged ischemia, lactic acidosis and associated proton generating pathways can cause intracellular pH to fall below 6.4 [47-49]. Here, to simulate the effects of in vivo graded ischemia we subjected cardiac myocytes to conditions previously established by our laboratory to mimic no-flow ischemia, progressive acidosis and caspase-dependent apoptotic cell death [10,11]. Under these conditions we found that miR-126 was induced by >4-fold and the apoptotic index increased to >25% after 72h of hypoxia/acidosis and a medium pH of 6.7. The increased level of apoptosis was accompanied by sustained inductions of inflammatory cytokines, TNFα, IL-6 and IL-8, elevated phosphorylation of p38-MAPK and JNK and decreased expression of anti-apoptosis Bcl-2. KD of miR- 126 by delivery of a selective antimir prior to hypoxia/acidosis, normalized expression of cytokines, eliminated the enhanced phosphorylation of p38-MAPK and JNK, significantly abrogated apoptotic cell death, and re-established control levels of Bcl-2.
Importantly we found that experimental manipulation of medium acidosis in parallel cardiac myocytes cultured aerobically in the absence of hypoxia reproduced almost identical results with respect to all measured parameters. This is consistent with our previous reports that hypoxia alone does not confer apoptosis without coincident acidosis or subsequent reoxygenation [9-11]. Indeed, we detected only minimal apoptosis even when cardiac myocytes were subjected to 6 days of pH neutral hypoxia [9]. Together the results support a pathway wherein miR-126 plays a central role in cardiac myocyte death caused by exposure to acidosis with or without concurrent hypoxia by regulating signaling pathways of inflammatory cytokine production, p38-MAKP/JNK and Bcl-2. The findings are consistent with our previous reports that hypoxia with concurrent acidosis, but not hypoxia alone confers lethal cardiac myocyte injury through BH3-only pathways [9-11,35,36,50]. The results are also consistent with our previous report, and that of others that miR-126, while generally considered to be endothelial-specific is induced by hypoxia in non-endothelial cells including cardiac myocytes and regulates pro-survival kinases and inflammatory cytokines [32,51]. Acidosis appears to be the critical regulator of miR-126 expression in this model and the implications of the studies extend beyond those of ischemic cardiac injury to other conditions of acidosis including rapidly growing solid tumors. However, the main clinical relevance is cardiac ischemia, a pathophysiological state that can include extreme acidosis.
Previous work has described both positive and negative roles for miR-126 in the regulation of angiogenesis, apoptosis and cell proliferation in different cells and tissues including the heart, tumors and retinal cells primarily by targeting protein kinase signaling pathways including PTEN/PI3K/Akt, mTOR, ERK/MAPK, Notch [52-60]. These studies document the diverse targets of miR- 126 and illustrate context-dependent actions with differential effects on cell survival and proliferation that may be determined in part by the metabolic state of the responding tissue [44,61,62]. As discussed in a recent review by Nammian et al. [44], miR-126 may confer opposite consequences depending on the biological context. Elevated miR-126 was shown to inhibit proliferation and activate apoptosis of human lung endothelial and HeLa cells respectively [41,43]. Context dependency was also reported in rats where upregulated miR-126 conferred increased levels of inflammatory factors and enhanced cardiomyocyte apoptosis, whereas simultaneous overexpression of E2F3 (a miR-126 target) with miR-126 had the opposite effect [55]. Our studies indicate increased expression of miR-126 in cardiac myocytes exposed to acidosis with or without hypoxia that promotes activation of p38- MAPK and JNK and increased production of pro-inflammatory cytokines TNFα, IL-6 and IL-8, suppression of Bcl-2 and consequent elevation of apoptosis. Whereas neonatal cardiac myocytes may not always fully reproduce the responses of adult myocytes to experimental manipulations, our previous work as well as that of others established that similar molecular pathways of injury by simulated ischemia with acidosis and/or reoxygenation is also seen in adult cardiac myocytes and intact heart as well as in astrocytes [63,64], but not cardiac fibroblasts [10], suggesting that it is a biologically relevant model.
Conclusions
We describe central roles for miR-126 in the responses of cardiac myocytes to acidosis whether the intracellular pH autoregulates in response to metabolic changes caused by chronic hypoxia or by experimental manipulation of extracellular pH. miR-126 levels were increased similarly by both conditions and paralleled by activations of inflammatory cytokines, stress kinases p38-MAPK and JNK, suppression of Bcl-2 and extensive apoptotic death of cardiac myocytes. Cause and effect of miR-126 was confirmed by demonstration that increased apoptosis and all signaling intermediates were abolished by KD of miR-126 with a selective antagomir.
Funding
Supported by 2012A032200005 (to JXL); R24EY028764 (to KAW), R43EY031238 (to KAW).
References
- Webster K A (2007) Programmed death as a therapeutic target to reduce myocardial infarction. Trends Pharmacol Sci 28(9): 492-499.
- Webster K A (2009) Mitochondrial Death Channels. Am Sci 97(5): 384-391.
- Patel P, Karch J (2020) Regulation of cell death in the cardiovascular system. Int Rev Cell Mol Biol 353: 153-209.
- Olivetti G, Abbi R, Quaini F, J Kajstura, W Cheng, et al. (1997) Apoptosis in the failing human heart. N Engl J Med 336(16): 1131-1141.
- Narula J, Haider N, Virmani R, T G DiSalvo, F D Kolodgie, et al. (1996) Apoptosis in myocytes in end-stage heart failure. N Engl J Med 335(16):1182-1189.
- Mallat Z, Tedgui A, Fontaliran F, R Frank, M Durigon, et al. (1996) Evidence of apoptosis in arrhythmogenic right ventricular dysplasia. N Engl J Med 335(16): 1190-1196.
- Saraste A, Pulkki K, Kallajoki M, K Henriksen, M Parvinen, et al. (1997) Apoptosis in human acute myocardial infarction. Circulation 95(2): 320-323.
- Fliss H, Gattinger D (1996) Apoptosis in ischemic and reperfused rat myocardium. Circ Res 79(5): 949-956.
- Webster KA, Bishopric NH (1992) Molecular regulation of cardiac myocyte adaptations to chronic hypoxia. J Mol Cell Cardiol 24(7): 741-751.
- Webster K A, Discher D J, Kaiser S, Olga Hernandez, Barbara Satoet al. (1999) Hypoxia-activated apoptosis of cardiac myocytes requires reoxygenation or a pH shift and is independent of p53. J ClinInvest 104(3): 239-252.
- Kubasiak L A, Hernandez O M, Bishopric N H, Keith A Webster (2002) Hypoxia and acidosis activate cardiac myocyte death through the Bcl-2 family protein BNIP3. Proc Natl Acad Sci USA 99(20): 12825-12830.
- Mahtta D, Sudhakar D, Koneru S, Guilherme Vianna Silva 3, Mahboob Alam, et al. (2020) Targeting Inflammation After Myocardial Infarction. Curr Cardiol Rep 22(10): 110.
- Andreadou I, Cabrera-Fuentes H A, Devaux Y, Nikolaos G Frangogiannis, Stefan Frantz, et al. (2019) Immune cells as targets for cardioprotection: new players and novel therapeutic opportunities. Cardiovasc Res 115(7): 1117-1130.
- Mauro A G, Bonaventura A, Mezzaroma E, Mohammed Quader, Stefano Toldo et al. (2019) NLRP3 Inflammasome in Acute Myocardial Infarction. J Cardiovasc Pharmacol 74(3): 175-187.
- Long X, Crow M T, Sollott S J, Enhanced expression of p53 and apoptosis induced by blockade of the vacuolar proton ATPase in cardiomyocytes, et al. (1998) Enhanced expression of p53 and apoptosis induced by blockade of the vacuolar proton ATPase in cardiomyocytes. J Clin Invest 101(6): 1453-1461.
- Karwatowska-Prokopczuk E, Nordberg J A, Li H L, R L Engler, R A Gottlieb, et al. (1998) Effect of vacuolar proton ATPase on pHi, Ca2+, and apoptosis in neonatal cardiomyocytes during metabolic inhibition/recovery. Circ Res 82(11): 1139-1144.
- Taylor C J, Nicola P A, Wang S, Margery A Barrand, Stephen B Hladky, et al. (2006) Transporters involved in regulation of intracellular pH in primary cultured rat brain endothelial cells. J Physiol 576(Pt 3): 769-785.
- Quade B N, Parker M D, Occhipinti R (2021) The therapeutic importance of acid-base balance. Biochem Pharmacol 183: 114278.
- Li J, Eastman A (1995) Apoptosis in an interleukin-2-dependent cytotoxic T lymphocyte cell line is associated with intracellular acidification. Role of the Na(+)/H(+)-antiport. J Biol Chem 270(7): 3203-3211.
- Pérez-Sala D, Collado-Escobar D, Mollinedo F (1995) Intracellular alkalinization suppresses lovastatin-induced apoptosis in HL-60 cells through the inactivation of a pH-dependent endonuclease. J Biol Chem 270(11): 6235-6242.
- Gottlieb R A, Giesing H A, Zhu J Y, R L Engler, B M Babior et al. (1995) Cell acidification in apoptosis: granulocyte colony-stimulating factor delays programmed cell death in neutrophils by up-regulating the vacuolar H(+)-ATPase. Proc Natl Acad Sci U S A 92(13): 5965-5968.
- Montgomery R L, van Rooij E (2011) Therapeutic advances in MicroRNA targeting. J Cardiovasc Pharmacol 57(1): 1-7.
- Calin G A, Croce C M (2006) MicroRNA signatures in human cancers. Nat Rev Cancer 6(11): 857-866.
- Villain G, Poissonnier L, Noueihed B, Gaëlle Bonfils 1, Jose Carlos Rivera, et al. (2018) miR-126-5p promotes retinal endothelial cell survival through SetD5 regulation in neurons. Development 145(1): dev156232.
- Fish J E, Santoro MM, Morton SU, Sangho Yu, Ru-Fang Yeh, et al. (2008) miR-126 regulates angiogenic signaling and vascular integrity. Dev Cell 15(2): 272-284.
- Wang S, Aurora A B, Johnson B A, Xiaoxia Qi, John McAnally, et al. (2008) The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell 15(2): 261-271.
- Long G, Wang F, Duan Q, Fuqiong Chen, Shenglan Yang, et al. (2012) Human circulating microRNA-1 and microRNA-126 as potential novel indicators for acute myocardial infarction. Int J Biol Sci 8(6): 811-818.
- Yu W, Chen J (2020) Circulating HOTAIR/miR-126 axis is negatively associated with disease risk of incident myocardial infarction. Int J Cardiol 298: 121.
- Chistiakov D A, Orekhov A N, Bobryshev Y V (2016) The role of miR-126 in embryonic angiogenesis, adult vascular homeostasis, and vascular repair and its alterations in atherosclerotic disease. J Mol Cell Cardiol 97: 47-55.
- Somanath PR, Razorenova OV, Chen J (2006) Akt1 in endothelial cell and angiogenesis. Cell Cycle 5(5): 512-518.
- Niu G, Chen X (2010) Vascular endothelial growth factor as an anti-angiogenic target for cancer therapy. Curr Drug Targets11(8): 1000-1017.
- Ren Y, Bao R, Guo Z, Jin Kai 1, Chen-Ge Cai, et al. (2021) miR-126-5p regulates H9c2 cell proliferation and apoptosis under hypoxic conditions by targeting IL-17A. Exp Ther Med 21(1):67.
- Wei L, Chen Z, Cheng N, Xing Li,1 Jie Chen, et al. (2020) MicroRNA-126 Inhibit Viability of Colorectal Cancer Cell by Repressing mTOR Induced Apoptosis and Autophagy. Onco Targets Ther 13: 2459-2468.
- Zhang Z, Wang J, Cheng J (2018) Effects of miR-126 on the STAT3 signaling pathway and the regulation of malignant behavior in lung cancer cells. Oncol Lett 15(6): 8412-8416.
- Graham R M, Frazier D P, Thompson J W, Shannon Haliko, Huifang Li, et al. (2004) A unique pathway of cardiac myocyte death caused by hypoxia-acidosis. J Exp Biol 207(Pt 18): 3189-3200.
- Webster KA, Discher DJ, Hernandez OM, K Yamashita, NH Bishopric, et al. (2000) A glycolytic pathway to apoptosis of hypoxic cardiac myocytes. Molecular pathways of increased acid production. Adv Exp Med Biol 475: 161-175.
- Dougherty C J, Kubasiak L A, Prentice H, Peter Andreka, Nanette H Bishopric, et al. (2002) Activation of c-Jun N-terminal kinase promotes survival of cardiac myocytes after oxidative stress. Biochem J 362(Pt 3): 561-571.
- Markou T, Dowling A A, Kelly T (2009) Regulation of Bcl-2 phosphorylation in response to oxidative stress in cardiac myocytes. Free Radic Res 43(9): 809-816.
- Webster K A, Discher D J, Bishopric N H (1994) Regulation of fos and jun immediate-early genes by redox or metabolic stress in cardiac myocytes. Circ Res 74(4): 679-686.
- Ebrahimi F, Gopalan V, Wahab R, Cu-Tai Lu 3, Robert Anthony Smith, et al. (2015) Deregulation of miR-126 expression in colorectal cancer pathogenesis and its clinical significance. Exp Cell Res 339(2): 333-341.
- Ichikawa R, Kawasaki R, Iwata A, Sayaka Otani, Eiji Nishio, et al. (2020) MicroRNA‑126‑3p suppresses HeLa cell proliferation, migration and invasion, and increases apoptosis via the PI3K/PDK1/AKT pathway. Oncol Rep 43(4): 1300-1308.
- Feng S D, Mao Z, Liu C, Nie Y-S, Sun B, et al. (2017) Simultaneous overexpression of miR-126 and miR-34a induces a superior antitumor efficacy in pancreatic adenocarcinoma. Onco Targets Ther 10: 5591-5604.
- Cao D, Mikosz A M, Ringsby A J, Kelsey C Anderson 4, Erica L Beatman, et al. (2020) MicroRNA-126-3p Inhibits Angiogenic Function of Human Lung Microvascular Endothelial Cells via LAT1 (L-Type Amino Acid Transporter 1)-Mediated mTOR (Mammalian Target of Rapamycin) Signaling. Arterioscler Thromb Vasc Biol 40(5): 1195-1206.
- Nammian P, Razban V, Tabei S (2020) MicroRNA-126: Dual Role in Angiogenesis Dependent Diseases. Curr Pharm Des 26(38): 4883-4893.
- Guo Y, Yan B, Tai S, Shenghua Zhou 3, Xi-Long Zheng, et al. (2020) PCSK9: Associated with cardiac diseases and their risk factors?. Arch Biochem Biophys 2020:108717.
- Webster K A, Graham R M, Thompson J W, Maria-Grazia Spiga, Donna P Frazier, et al. (2006) Redox stress and the contributions of BH3-only proteins to infarction. Antioxid Redox Signal 8(9-10): 1667-1676.
- Handley M G, Medina R A, Nagel E (2011) PET imaging of cardiac hypoxia: opportunities and challenges. J Mol Cell Cardiol, 51(5): 640-650.
- Kette F, Weil M H, von Planta M, R J Gazmuri, E C Rackow, et al. (1990) Buffer agents do not reverse intramyocardial acidosis during cardiac resuscitation. Circulation 81(5): 1660-1666.
- Wu D, Kraut J A (2014) Role of NHE1 in the cellular dysfunction of acute metabolic acidosis. Am J Nephrol 40(1): 36-42.
- Frazier D P, Wilson A, Graham R M, John W Thompson, Nanette H Bishopric, et al. (2006) Acidosis regulates the stability, hydrophobicity, and activity of the BH3-only protein Bnip3. Antioxid Redox Signal 8(9-10): 1625-1634.
- Shi H, Chen L, Wang H, Shoukang Zhu, Chunming Dong, et al. (2013) Synergistic induction of miR-126 by hypoxia and HDAC inhibitors in cardiac myocytes. Biochem Biophys Res Commun 430(2): 827-832.
- Ge R, Lv Y, Li P, Lin Xu 1, Xiaoya Feng, et al. (2021) Upregulated microRNA-126 induces apoptosis of dental pulp stem cell via mediating PTEN-regulated Akt activation. J Clin Lab Anal 35(2): e23624.
- Norbeck T B (1998) The Connecticut State Medical Society Medical Courtesy Card Program. Conn Med 52(6): 364-365.
- Bi X, Lv X, Liu D, Guang Yao, Lijuan Wang, et al. (2020) METTL3-mediated maturation of miR-126-5p promotes ovarian cancer progression via PTEN-mediated PI3K/Akt/mTOR pathway. Cancer Gene Ther 28(3-4): 335-349.
- Xu Y (2020) TET2 expedites coronary heart disease by promoting microRNA-126 expression and inhibiting the E2F3-PI3K-AKT axis. Biochem Cell Biol 98(6): 698-708.
- Kong R, Gao J, Ji L (2020) MicroRNA-126 promotes proliferation, migration, invasion and endothelial differentiation while inhibits apoptosis and osteogenic differentiation of bone marrow-derived mesenchymal stem cells. Cell Cycle 19(17): 2119-2138.
- Wang Y, Wang M, Yu P, Li Zuo, Qing Zhou, et al. (2020) MicroRNA-126 Modulates Palmitate-Induced Migration in HUVECs by Downregulating Myosin Light Chain Kinase via the ERK/MAPK Pathway. Front Bioeng Biotechnol 8: 913.
- Kong Z, Wang Y, Zhang Y, Wei Shan, Jianping Wu, et al. (2020) MicroRNA-126 promotes endothelial progenitor cell proliferation and migration ability via the Notch pathway. Cardiovasc Diagn Ther 10(3): 490-499.
- Lou Z, Li Q, Wang C (2020) The effects of microRNA-126 reduced inflammation and apoptosis of diabetic nephropathy through PI3K/AKT signalling pathway by VEGF. Arch Physiol Biochem 2020: 1-10.
- Wei Y, Nazari-Jahantigh M, Neth P (2013) MicroRNA-126, -145, and -155: a therapeutic triad in atherosclerosis? Arterioscler Thromb Vasc Biol 33(3): 449-454.
- Li J, Yang C, Wang Y (2021) miR‑126 overexpression attenuates oxygen‑glucose deprivation/reperfusion injury by inhibiting oxidative stress and inflammatory response via the activation of SIRT1/Nrf2 signaling pathway in human umbilical vein endothelial cells. Mol Med Rep 23(2): 1.
- Gai H Y, Wu C, Zhang Y (2019) Long non-coding RNA CHRF modulates the progression of cerebral ischemia/reperfusion injury via miR-126/SOX6 signaling pathway. Biochem Biophys Res Commun 514(2): 550-557.
- Bodyyak N, Rigor DL, Yee-Shiuan Chen, Yuchi Han, et al. (2007) Uncoupling protein 2 modulates cell viability in a adult rat cardiomyocytes. Am J Physiol Heart Circ Physiol H829-835.
- Regula KM, Ens K (2002) Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-medicated cell death of ventricular myocytes. Circ Res: 226-231.
