 Research Article
Research ArticleAbstract
Beta-glucuronidase is an important enzyme involved in the digestion, particularly in phase 2 of liver detoxification especially in detoxifying estrogen like hormones and environmental toxins. Taking this into consideration, the present study was designed to select and identify potential scaffolds followed by beta-glucuronidase inhibition and computational studies. Six different types of scaffolds were selected based on their potential to interact therapeutically with beta-glucuronidase enzyme. Among them, the most potent compound was subjected to molecular docking studies and the results revealed significant interactions with important amino acid residues within the active site of beta-glucuronidase. Some compounds identical to marketed inhibitors of betaglucuronidase were retrieved from PubChem and Zinc database, and a defined library was generated. The pharmacophore method was applied for the screening of potent inhibitor of beta-glucuronidase. Structural similarity search feature assembled a total of >500 compounds. Out of which 9 compounds were selected and filtered out applying the already reported methods. These investigations can be used in the design of selective β-glucuronidase inhibitors for future studies. This analysis may lead to findings for novel β-glucuronidase inhibitors against digestion related disorders after some more advanced studies.
Keywords: Beta-Glucuronidase; Library Design; Inhibitory Efficacy; Virtual Screening; Molecular Docking
Introduction
Glycosidase Family of Enzymes
Glycosidases (GH) are vital for almost all living organisms (exceptions are some Archaeans and a few unicellular parasitic eukaryotes) playing diverse and different roles. Considering the diversity of reactions, they catalyze, amino acid sequence and folding, glycosidases have been classified in many different categories [1].
Beta-Glucuronidases
Beta-glucuronidase (β-glucuronidase) enzyme (EC3.2.1.31) is a glycosidase that have role in the hydrolysis of glycosaminoglycans at specific glucuronide site [2]. These specific enzymes are found in plants, bacteria and vertebrates. Intestinal β-glucuronidase is categorized as one of the key enzymes for the treatment of numerous intestinal diseases. In invertebrates, β-glucuronidase from mollusk sp (Ampullaria and Helix pomatia) are used in the hydrolysis of steroids in order to assess urinary conjugate cortisol [3]. Besides intestinal β-glucuronidase, the lysosomal β-glucuronidase have been found active in acidic condition (pH ~ 4-5). It has also been observed that there is a direct effect of pH and temperature on the stability of enzyme. The stability of β-glucuronidase is determined and confirmed by residual activity at varying temperature of standard (37 °C) and optimum temperature (70 °C). At both temperatures, the enzymes have a positive response from acidic to neutral pH. At 75 °C temperature and pH above 8, the activity of enzyme is lost [3]. Other key roles of β-glucuronidase includes carbohydrate processing. temperatures, the enzymes have a positive response from acidic to neutral pH. At 75 °C temperature and pH above 8, the activity of enzyme is lost [3]. Other key roles of β-glucuronidase includes carbohydrate processing.
They also show functional importance and are used in the hydrolysis of various steroids, bioassay for plant hormones, prodrug therapy [4] and as biomarker [5] and are present in extracellular fluid during the inflammation [6]. In human plasma, β-glucuronidase serves as a biomarker for the identification of human exposure to organophosphate [4]. The deficiency of this enzyme can lead to autosomal recessive disorders which is mucopolysaccharidosis VII, commonly known as Sly syndrome [4]. Genetic studies have revealed that β-glucuronidase gene (GUSB) plays an important role in various applications of plant biotechnology. β-Glucuronidase proteins have also been developed by cloning, expression and purification of enzymes (enzyme replacement therapy). The protein, isolated from most vertebrates, occurs in the form of homotetramers with a molecular mass of around 280-300 KDa while the lysosomal β-glucuronidase purified by various chromatographic techniques possesses a molecular mass of 50 KDa [7].
Structure of Beta-Glucuronidases
Human beta-glucuronidase is found to be as 80 kDa in monomeric form (653 amino acids residues) and its proteolysis cleaves 18 amino acid residues from the carboxy terminal end leaving it as 78 kDa monomer. It exists in homotetrameric form with 332 kDa while carrying various notable structural changes, including beta barrel type, also known as a jelly-roll barrel, and the TIM barrel [8]. The crystal structure of the enzyme has been solved by X-ray crystallography. The specific site for the transport of lysosomal β-glucuronidase is mannose 6-phosphate receptor (MPR) [9].
Chromosomal Location of Beta-Glucuronidases GAUSB Gene
Its cytogenetic location is 7q11.21, on the long (q) arm of chromosome, number 7 at 11.21 position (Figure 1) while its molecular location is 65,960,684 to 65,982,314 base pairs on chromosome number 7 (Homo sapiens Annotation Release 109, GRCh38.p12) (NCBI).
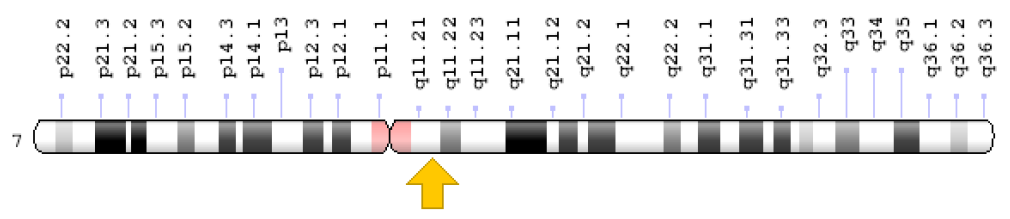
Figure 1: Image of melanoma (a) and nevus tissues (b). Source: ISIC 2019.
Beta-glucuronidase and Diseases
In 2000, Sperker et al. addressed the least investigated issue regarding the excess release of beta-glucuronidase in pancreatic cancer [10]. This fatal pancreatic cancer is world’s 5th cause of death. This study illustrated the excess release of enzyme by carcinoma cells as compared to normal cells. It is also up regulated in different pathological diseases like urinary tract infection and renal diseases. It also causes transplant rejection, epilepsy and cancers i-e., breast and larynx cancer. Moreover, it is involved in the bone related disorders like inflammatory joint disorder and rheumatoid arthritis, and several hepatic diseases, and later acquired immunodeficiency syndrome which is linked to the overexpression of beta-glucuronidase [11]. In many tumors and inflammatory areas, β-glucuronidase activity is high and the enzyme is secreted into the extracellular space [10]. Since then, the researchers started focusing the inhibition of beta-glucuonidase enzyme through the development of various inhibitors mainly nitrogen heterocyclic compounds such as indole, triazoles and benzothiazole [12].
Computational Screening
Virtual screening (VS) is a powerful computer-based technique which has widely been used for the identification of promising compounds such as specific inhibitors that are capable of binding with known molecular target structures. VS is considered as an imperative technique, for new inhibitors and molecular drug discovery, depending upon the growing demand of protein and nucleic acid structure. Virtual screening evaluates numerous compounds all at once, two significant steps should be well understood and perceived for this purpose. Primary concern is to find docking technique which will enable us to acquire our desired configuration [13]. To justify a docking method, various experiments of redocking are accomplished, where separation of whole series of known complexes are completed followed by the process of redocking. The secondary concern is to assure whether this docking strategy will be able to replicate the detected binding mode. Through these validation studies, nearly 10 degree of torsional freedom was found in drug like molecules of recent version of Auto-Dock. Furthermore, next step is to ensure experimentally that the compounds do bound at their predicted binding energy, allowing precise grading of compounds [13].
Solely standard deviation of 2-3 Kcal/mol, provides efficiency of energy prediction through computational based docking algorithms and Auto Dock which is insufficient for promising ranking though. The compound enrichment process is implied where predicted compounds bind tightly inferring to give away real solid bonding upon testing experimentally. In this methodology of two-step docking, Auto-Dock yields more for virtual screening, because at the start of screening interaction map is calculated only once where further working on docking simulation takes place. Expansion of Auto Dock can be accomplished through various map modification methods. Prior to docking, grid maps are modified for contemporary chemical or physical property insertions, Example are maps for predicting covalently linked complexes, covalent maps; maps for bridging water position prediction, water maps; for de novo design of ligand, mutable atom approach; and for modelling protein flexibility, energy weighted averages [14].
Docking Based Screening (DBVS)
The workflow chart for docking-based virtual screening (DBVS) presented below functions normally using targeted structure for input, which can be modelled experimentally or on computer basis together with small-molecules containing compound library which is either purchased or synthesized (Figure 2). Proper assignment of protonation, tautomeric, and stereoisomeric states are also the preparations required for both target and compound libraries. Using docking program, every compound of the library is docked into its target binding site virtually. An optimum amount of complementarity of steric and physicochemical properties by computationally modelling the ligand-target interaction are achieved using this program. Subsequently, a mathematic algorithm “Scoring Function” is signed to assess the accuracy level among target and the docked compound followed by ranking and selection, which is put through by binding score calculation, only some of them from a group of top-ranked compounds were selected to perform experimental assay [13,14].
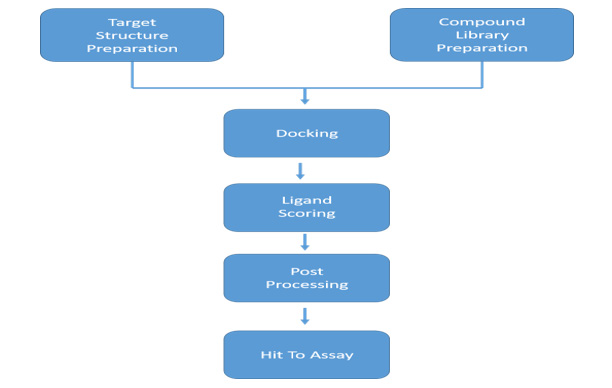
Figure 2: A simple working mechanism for docking-based virtual screening (DBVS).
Absence of suitable accessible database containing purchased compounds is the most common obstruction faced during data entry into SBVS. PubChem databases have a library of 609 molecules including their 3D structures. Therefore, PubChem database have prepared a library of 609 molecules including their 3D structures by engaging the catalogues from the vendors, thus broadening the library size repeatedly. Molecules have been assigned with protonation properties like Log P value, rotatable bond’s number and molecular weight beforehand, along with complete vendor and purchasing information. Manifold configurations are available in one format, open option of free download in numerous formats like SMILES, mol2, 3D SDF and DOCK FleX is also available at this database (http://zinc.docking.org). Other query tools for direct insertion and search are also accessible accompanied by user choice of uploading their own molecules [15].
Auto Dock Vina efficiency can be noticed from the fact that it can acquire two orders of magnitude speed up with better binding mode prediction efficiency as compared to old version of molecular docking software Auto Dock 4. Moreover, parallelism is accompanied for speeding-up further with the help of multithreading technique through multi-core machines. This software provides transparent results directly to user, after mechanically computing grid maps and clusters [16]. Two approaches were used to split VS, hinge on target and current ligands information retrieve in the start of screening, into SBVS and LBVS. LBVS strategy, for identification of known active or inactive compound looks into biological data. The main purpose of this data is retrieval of other efficiently active molecular platforms depending on common descriptors values or similarity measures. None of any strategies exceeds one another; however, they can be used both-ways either collectively or individually. Advance research system promoted the idea of unified approach of combining the two: LBVS & SBVS either in parallel or sequential manner, because of its notable prospective of exploiting the whole structural and chemical information available during the hunt for novel molecules. Its availability is dependent upon the presence of known active compounds together with the 3D structural protein targets such as X-ray, NMR, comparative modelling. As compared to parallel combination, serial is more of a hierarchical and rapid in the meantime less advanced method. Unlike serial, parallel combination focuses on juxtaposition of selected compounds, by utilizing either complementary selection considering top-ranked from each method only or by consensual selection including the compounds of both approaches [17].
In-Silico Study
Library Retrieval: The software Chem Draw is developed by Cheminformatics Company, Cambridge soft (cambridgesoft. com) for performing molecular editing. After downloading these compounds with their respective zinc and PubChem ID they were drawn on molecule editor, computational chemical drawing software (http://zinc.docking.org) [15]. The similar chemical structures of standard inhibitor for beta-glucuronidase (GusB) [18] from ZINC and PubChem were downloaded which were 500 [15]. Subsequently, all the structures were drawn using a computational tool Chem Draw and saved as cdx files.
Pharmacophore Formation: It is a 3-dimensional (3D) array of the chemical features which are included in the activation or inhibition of enzyme/receptor. The concept is often used to develop methods for computer aided designs for drugs. It involves the 3D arrangement of ligand features that take part in action against targeting protein. MOE software was used to generate pharmacophore model [19].
MOE Pharmacophore Tools: The Extended Hueckel Theory (EHT) pharmacophore modelling is based on the semi-empirical approach for the features and annotations of pharmacophore [20]. This approach uses ligand resonance and electron withdrawing effects and therefore the pharmacophore features were formed by this theory are sensitive to non-standard interactions, like carbon to hydrogen and halogen bonding interactions, during this pharmacophore screening. The usefulness of this approach includes:
a) SMARTS patterns are not required for annotations.
b) Resonance effects are captured in the acceptor and donor
strengths.
c) Each feature has an associated strength value, so it is easy
to differentiate between weak and strong features.
d) Weak-weak interactions are avoided.
e) New pharmacophore features through C-H and halogen
bond interactions.
This explores the following concepts:
1) A beta-glucuronidase (GusB) inhibitor model is drawn
using EHT Scheme.
a) Creating a pharmacophore query
b) Preparing the system
c) Creating a Don feature
d) Creating an Acc feature
e) Creating Aro feature
f) Creating a Don2 feature
g) Saving a pharmacophore query
h) Creating excluded volume around pocket atoms
i) Running a pharmacophore search
2) Analyzing the output
3) Molecular Docking
Substrates and inhibitors that have the potential to become
a novel drug are mostly discovered by accident in systematic
screening by using a trial-and-error method. In vitro experiments
that are extremely tedious and very costly are used to test the
action of many compounds to the target/protein. If the target 3D
structure is available, then it is capable for molecular docking for
the process of identification for the drug or inhibition quality [21].
Standard Precision (SP) and Extra Precision (XP) Docking: It is fundamentally a structural sampling process in many docked structures to find the one with the potential, which is subsequently subjected to experimental NMR spectroscopic and X-ray crystallographic analysis. The sampling process for structures runs on genetic algorithm that stimulate Annealing, Monte-Carlo simulation, the distance geometry and miscellaneous methods, following the energy function which is used to find fitness and accuracy in target protein and ligand. Final docked structures are nominated on the basis of energy function. Low scoring represents the fair binding between ligand and protein [22]. Molecular docking method uses full energy function approach for the ranking of the protein-ligand structures. The main goal is to predict the binding site on receptor protein and to understand the binding-binding genomics of the ligand. Major challenges faced in computer aided docking are namely and appropriate ligand-receptor binding flexibility and realistic prediction of ligand-receptor binding energies [23].
Binding Energy: Computer-aided docking targets the prediction of good assessment for binding energies of ligandreceptor with more efficiency, it uses many docking programs. The docking program we used is Lead IT [21] which combines main application areas of modern-early discovery of drug, its users can now have a very user-friendly graphical interface that sets in award-winning as well as long-standing molecular docking alongside advanced fragment-based design tools: FlexX and Re Core. FlexX is completely auto molecular docking program available on Lead IT 2.1.6 package and was used for docking compounds into the active sites of enzymes. FlexX is considered ligand flexible due to the changing of structures of ligand in active site while making receptor site in the protein rigid [21,24].
Visualization: Discovery Studio Visualizer v4 was used for the visualization of 3D putative binding modes acquired from docking [25].
Result and Discussion
To find the potent inhibitor of beta-glucuronidase, 6 previously known inhibitors including benzothiazole, castanospermine, mucate, saccharate, saccharic acid and silymarin were screened in silico. From PubChem total similar structures were 500. After virtual screening of compounds, nine were shortlisted which have the highest binding energies. Among them, three have similar structures of benzothiazole, two are of castanospermine, one of mucate, two of saccharic acid and one of silymarin. Benzothiazole similar compound of ID number 128635 have the highest binding energy ‒32.21. Its molecular weight is 322.43 g/mol and its IUPAC name is 1,3-benzothiazol-6-yl-(4-piperidin-1-ylphenyl) diazene (Table 1). Pharmacophore studies were performed for the prediction of ADME (absorption, distribution, metabolism, and excretion) profile on all the nine compounds and their physicochemical parameters as shown in Table 1. The resulting structures have the same interactions of hydrogen bonds, salt bridges, hydrophobic interactions and metal interactions as of similar to the MRD in 3HN3 protein ((4R)-2-methylpentane-2,4-diol). 3HN3 is a human beta-glucuronidase at 1.7 Å resolution (Figure 3).
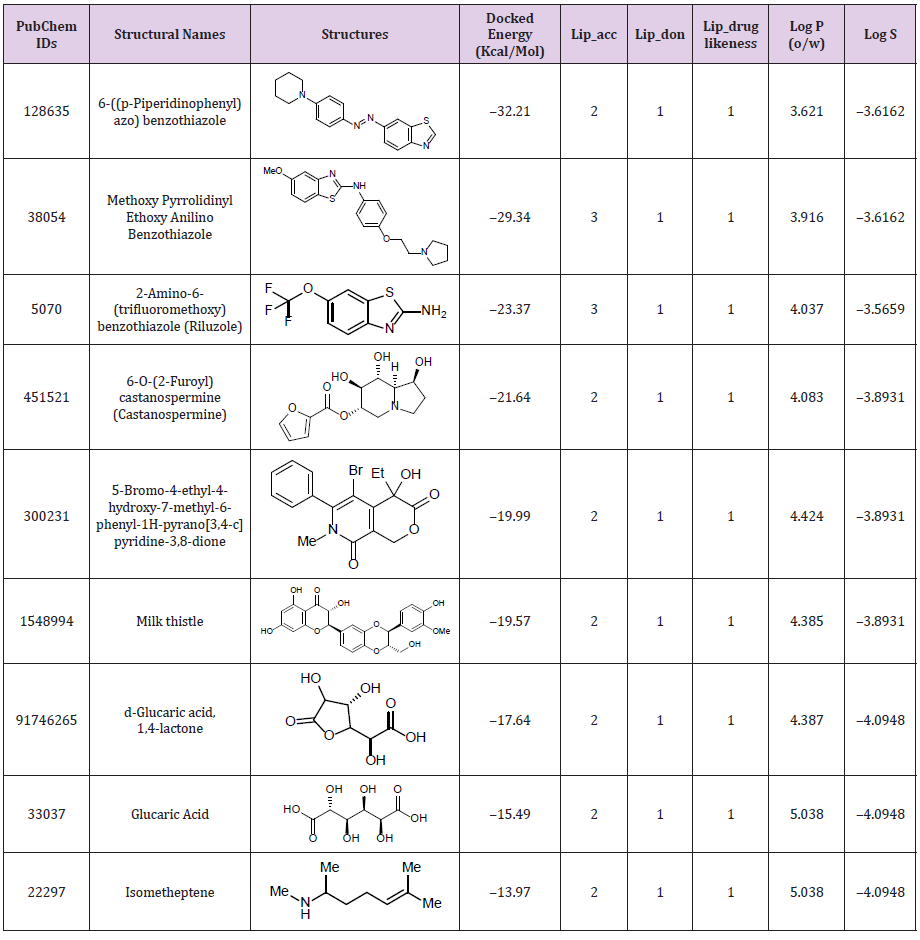
Table 1: Docking energies and physicochemical parameters of compounds after pharmacophore screening used for the prediction of ADME profile.
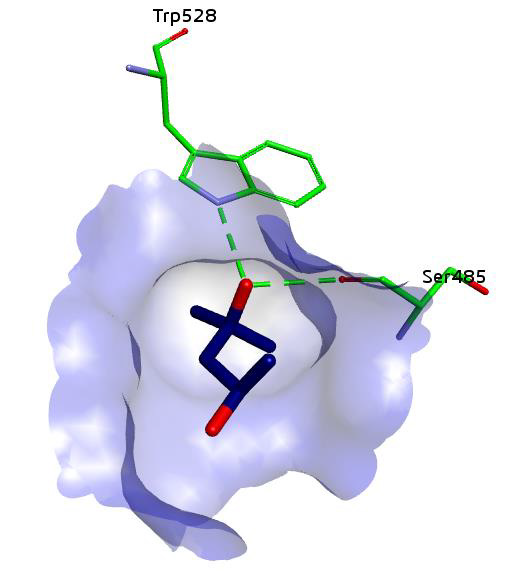
Figure 3: Putative binding of ligand (MRD) in beta-glucuronidase.
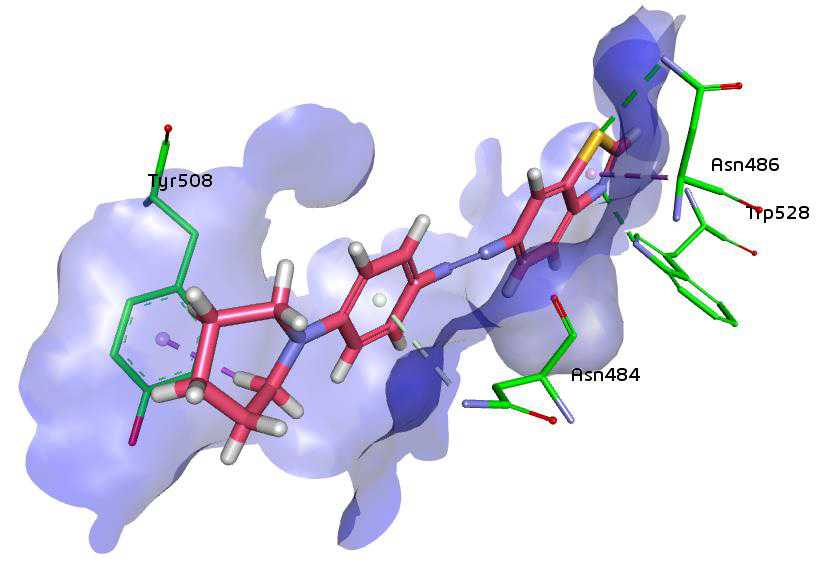
Figure 4: Binding of compound 1,3-benzothiazol-6-yl-(4-piperidin-1-ylphenyl) diazene in beta-glucuronidase showing hydrogen bonding and hydrophobic interactions.
Same interactions were found in the most potent inhibitor of the series which is 1,3-benzothiazol-6-yl-(4-piperidin-1-ylphenyl) diazene. All the interaction of this compound are showed in the given 3D structure which is generated through the discovery studio. The resulting image depicts the position of the compound inside the active pocket of beta-glucuronidase. Specificity pocket formed by the residues Trp528, Asn484, Ser485, Asn482, His509 and Tyr508 occupies by the benzothiazole ring (Figure 4).
Conclusion
Beta-glucuronidase inhibition offers potential therapeutic target for the management of digestive tract related diseases. Keeping this in view, the present study was designed to select and identify potential scaffolds from various classes of compounds followed by beta-glucuronidase inhibition and computational studies. Compounds identical to cognate ligand of human betaglucuronidase were retrieved from PubChem database and library was generated. The pharmacophore method was applied for the screening of potent inhibitor of beta-glucuronidase. From total 500 compounds, 9 compounds were selected and filtered out applying the above-mentioned method. The compounds were finally scrutinized based on binding energy and interactions with the active site residues. The highest binding energy was found to be for PubChem ID 128635, among benzothiazole based compounds as ‒32.21. Physicochemical parameters of compounds, selected after pharmacophore generation were used for the prediction of ADME profile.
Conflict of Interest
The authors declare no conflict of interest, financial or otherwise.
Acknowledgement
We acknowledge the support of Department of Biochemistry, University of Central Punjab, Lahore, Pakistan.
References
- Henrissat B (1998) Glycosidase families. Biochemical Society Transactions 26(2): 153-156.
- Konada RSR, Venugopal A, Nadimpalli SK (2020) Purification, biochemical and biophysical characterization of lysosomal β-D-glucuronidase from an edible freshwater mussel, Lamellidens corrianus. International Journal of Biological Macromolecules 152: 465-472.
- Rabbani G, Kaur J, Ahmad E, Khan RH, Jain S (2014) Structural characteristics of thermostable immunogenic outer membrane protein from Salmonella enterica serovar Typhi. Applied Microbiology and Biotechnology 98: 2533-2543.
- Ueyama J (2018) Human biomonitoring as a useful approach to health risk assessment compared with occupational exposure assessment of insecticide intake: Fundamental study focused on local populations and occupational fields. Nihon eiseigaku zasshi. Japanese Journal of Hygiene 73: 247-256.
- Chilke AM (2010) Kinetic study of hepatic β-glucuronidase in the Indian major carp, Labeo rohita (Hamilton). Fish Physiology and Biochemistry 36: 1145-1149.
- Markantes GK, Michalaki MA, Vagenakis GA, Lamari FN, Pitsi E, et al. (2019) Plasma beta-glucuronidase activity: a novel tool to distinguish type 1 from type 2 amiodarone-induced thyrotoxicosis? European Thyroid Journal 8: 215-220.
- Zhang Q, Gao B, Xiao Y, Yang H, Wang Y, et al. (2020) Purification and characterization of a novel β-glucuronidase precisely converts glycyrrhizin to glycyrrhetinic acid 3-O-mono-β-D-glucuronide from plant endophytic Chaetomium globosum DX-THS3. International Journal of Biological Macromolecules 159: 782-792.
- Kim HW, Mino K, Ishikawa K (2008) Crystallization and preliminary X-ray analysis of endoglucanase from Pyrococcus horikoshii. Acta Crystallographica Section F: Structural Biology and Crystallization Communications 64(12): 1169-1171.
- Bhamidimarri PM, Krishnapati LS, Ghaskadbi S, Nadimpalli SK (2018) Mannose 6‐phosphate‐dependent lysosomal enzyme targeting in hydra: a biochemical, immunological and structural elucidation. FEBS Letters 592: 1366-1377.
- Sperker B, Werner U, Mürdter TE, Tekkaya C, Fritz P, et al. (2000) Expression and function of β-glucuronidase in pancreatic cancer: potential role in drug targeting. Naunyn-Schmiedeberg's Archives of Pharmacology 362(2): 110-115.
- Taha M, Ismail NH, Jamil W, Khan KM, Salar U, et al. (2015) Synthesis and evaluation of unsymmetrical heterocyclic thioureas as potent β-glucuronidase inhibitors. Medicinal Chemistry Research 24(8): 3166-3173.
- Baharudin MS, Taha M, Imran S, Ismail NH, Rahim F, et al. (2017) Synthesis of indole analogs as potent β-glucuronidase inhibitors. Bioorganic Chemistry 72: 323-332.
- Cheng T, Li Q, Zhou Z, Wang Y, Bryant SH (2012) Structure-based virtual screening for drug discovery: a problem-centric review. The AAPS Journal 14(1): 133-141.
- Cosconati S, Forli S, Perryman AL, Harris R, Goodsell DS, et al. (2010) Virtual screening with AutoDock: theory and practice. Expert Opinion on Drug Discovery 5(6): 597-607.
- Irwin JJ, Shoichet BK (2005) ZINC - A free database of commercially available compounds for virtual screening. Journal of Chemical Information and Modeling 45(1): 177-182.
- Trott O, Olson AJ (2010) Auto Dock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry 31(2): 455-461.
- Lavecchia A, Di Giovanni C (2013) Virtual screening strategies in drug discovery: a critical review. Current Medicinal Chemistry 20(23): 2839-2860.
- http://www.rcsb.org/structure/3HN3
- (2019) Chemical Computing Group's Molecular Operating Environment (MOE) MOE 2019.
- Rahman SU, Ali HS, Jafari B, Zaib S, Hameed A, et al. (2020) Structure-Based Virtual Screening of Dipeptidyl Peptidase 4 Inhibitors and their In vitro Analysis. Computational Biology and Chemistry 107326.
- (2017) Lead IT version 2.3.2; Bio Solve IT GmbH, Sankt Augustin, Germany.
- Trott O, Olson AJ (2010) Auto Dock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry 31(2): 455-461.
- Lavecchia A, Di Giovanni C (2013) Virtual screening strategies in drug discovery: a critical review. Current Medicinal Chemistry 20(23): 2839-2860.
- Schneider N, Lange G, Hindle S, Klein R, Rarey M (2013) A consistent description of hydrogen bond and dehydration energies in protein-ligand complexes: methods behind the HYDE scoring function. Journal of Computer-Aided Molecular Design 27: 15-29.
- (2019) BIOVIA Discovery Studio Client v19.1.0.18287. Accelrys Discovery Studio. Accelrys Software Inc, San Diego, USA.
