 Case Report
Case ReportAbstract
The skeletal muscle channelopathies represent a rare group of neuromuscular
disorders which are caused by genetic mutations regarding voltage-gated ion
channels, which play an important role in muscle membrane depolarization. Muscle
channelopathies are broadly divided into 2 main categories: nondystrophic myotonias
(NDM) and periodic paralysis (PP). Periodic paralysis (PP) are rare autosomal dominant
neuromuscular disorders, characterized clinically by periodic attacks of muscle weakness
in concomitance with serum potassium level alterations. It is possible to distinguish
normokalemic, hyperkalemic and hypokalemic paralysis. PP is caused by genetic
mutations in voltage-gate ion channels like sodium, potassium, and calcium channels,
which are determinant for muscle membrane depolarization. The most common genes
involved in pathogenesis are CACN1S, SCN4A and KCNJ2, encoding calcium, sodium, and
potassium channels (Table 1). Moreover, paralysis related to serum potassium values
may also occur in thyreotoxicosis [1], Liddle syndrome, Gitelman syndrome, primary
hyperaldosteronism, and acid-base balance disorders. In this case report, we describe a
case of transient paralysis and muscular weakness of both upper and lower limbs after
a high carbohydrate meal on the day before clinical presentation. Potassium level was
normal at the hospital admission, while high levels of creatine phosphokinase, (CPK),
Myoglobin, and Aspartate Aminotransferases (AST) was observed.
A missense mutation in CACNA1S exon 11 was identified. This case supports the
importance of a correct family history, muscular enzyme analysis and genetic study in a
normokalemic presentation of periodic paralysis.
Keywords: Hypokalemia; Periodic Paralysis; Transient Paralysis; Muscular Weakness; Muscular Pain; Creatine Phosphokinase; CACNA1S Gene
Abbreviations: Normal PP: Normokalemic Periodic Paralysis; HyperPP: Hyperkalemic Periodic Paralysis; HypoPP: Hypokalemic Periodic Paralysis; n.v.: Normal Value; EEG: Electroencephalogram
Background
Muscular channelopathies may have two different clinical manifestations: sudden attacks of flaccid transient paralysis, often associated with abnormal serum potassium levels (periodic paralysis) and muscular stiffness (myotonia) [2,3]. In channelopathies mutations commonly involve calcium, chlorine, sodium, and potassium channels. They have autosomal dominant inheritance. The gene loci include CACN1AS (calcium channel) [4,5] located on chromosome 1, CLCN1 (chlorine channel) [1,6] on chromosome [7] SCN4A [4,5,7,8] and KCNJ2 [9] (sodium and potassium channels, respectively) on chromosome 17. Hypokalemic periodic paralysis is a rare disorder with a prevalence of 1:100.000 [1] and a large spectrum of penetrance which is expressed in a wide variety of correlation between genotype and phenotype [10]. Moreover, the quality of life in these patients can be impaired for a chronic and progressive muscular dystrophy [11,12]. Periodic paralysis is usually classified on clinical and laboratory criteria (serum potassium values and response to potassium administration) and on genetic criteria. Reliyng on serum potassium levels, periodic paralysis is classified in mormokalemic (NormoPP), hyperkalemic (HyperPP) and hypokalemic (HypoPP) [1,2]. HyperPP, NormoPP and 10% cases of HypoPP are related to SCN4A mutations7, 70% cases of HypoPP are related to CACN1AS mutations [3,4], on the other hand KCNJ2 mutations are decisive for Andersen-Tawill Syndrome [9,12].
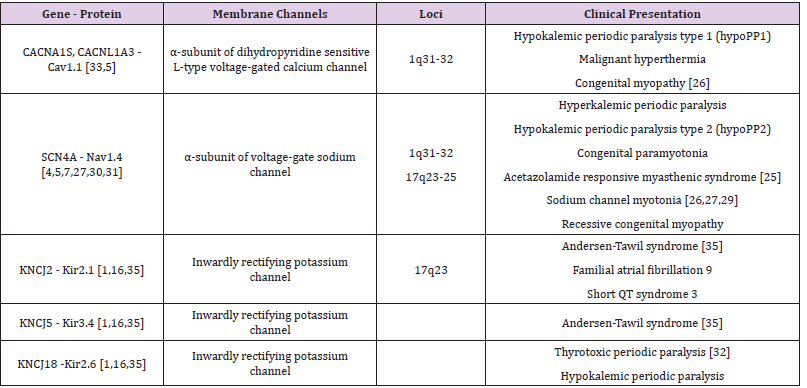
Table 1: Main channelopathies and loci involved. In channelopathies mutations interested calcium, sodium and potassium channels. The gene loci involved are CACN1AS (calcium channel) located on chromosome 1, SCN4A and KCNJ2 (sodium and potassium channels, respectively) located on chromosome 17. The table describes different clinical presentations related to specific gene loci mutations [28,29].
Clinically, HypoPP begins on the first or second decade and decrease in frequency after forty years of age [13].
Attacks are characterized by flaccid paralysis, usually
occurring on awakening during the night or at early-morning,
focal or generalized weakness which lasts for hours (occasionally
days) with a gradual resolution [1,2,14,15]. Occurrence may be
spontaneous or provoked by prolonged rest after vigorous exercise,
carbohydrate-rich meal on the previous day. Triggers include viral
infections, sleep deprivation, period in women and drugs (e.g. beta
agonists, corticosteroids and insulin) [1,16]. HyperPP differs from
HypoPP for an earlier age of onset (first decade) [1,16]. Weakness
is usually generalized and last 1-4 hours (infrequently days). They
are triggered by rest after exercise, K-rich foods, stress, and fatigue.
Among episodes of weakness, eyelid myotonia may be the only
clinical sign. Electrical myotonia is found in 50-75% of patients.
Typically, hyperkalemic attacks are associated with elevated ictal
serum potassium level, but many children have normal potassium
level during attacks. For those reason, laboratory approach is
important to distinguish them [1,16,17].
In our patient a transient paralysis and muscular weakness
with elevated levels of creatine phosphokinase, (CPK), Myoglobin,
and Aspartate Aminotransferases (AST), concurrently with normal
potassium serum levels was observed.
Case Presentation
A 12-years old Caucasian girl was admitted to our clinic unit
with acute onset of transient paralysis of all four limbs arisen on the
awakening and the followed by paresthesia’s, motor impairment
without loss or disturbances of consciousness [1,16]. There was also
muscular pain in both legs and arms without dysphagia, dysphonia,
history of suggestive symptoms such as ocular, sensory, cerebellar,
or cranial nerve involvement. Moreover, these symptoms improved
after salt supplement intake. Family history was suggestive for
muscle symptoms (not confirmed Liddle syndrome) [18]. Her
grandfather, father and one of her uncles had similar mild paralytic
attacks; on the other hand, another uncle presented more severe
symptoms, including dyspnea and respiratory muscles involvement.
She had a significant past medical history for transient paralysis.
One episode occurred after an acute diarrhea and two of them
during physical exercise with an improvement after few hours.
On examination, skin was not affected by cutaneous alterations or
lesions. Cardiovascular, pulmonary, and abdominal examinations
were not compromise [19]. Neurological exam revealed a normal
deep tendon reflexes and global and segmental strength of both
upper and lower extremities [1,16,20]. There were not palpable
goiter or tremors.
Laboratory findings of our case are shown in (Table 2). No
ECG alterations was recorded [19,20]. Arterial pressure levels
control were normal as well as the electroencephalogram. During
hospitalization, the girl never showed neurological or muscular
symptoms, so we prescribed oral potassium supplement only
in case of need [21]. We planned a mutation screening for
hypokalemic periodic paralysis. Genomic DNA was extracted from
the peripheral blood and also other family members are tested with PCR amplification and direct sequencing of CACNA1S exon 11, the
most frequent gene involved in hypokalemic periodic paralysis.
This mutation has been found in our patient and also in her father,
in two uncles, grandfather and an asymptomatic cousin (Figure 1).
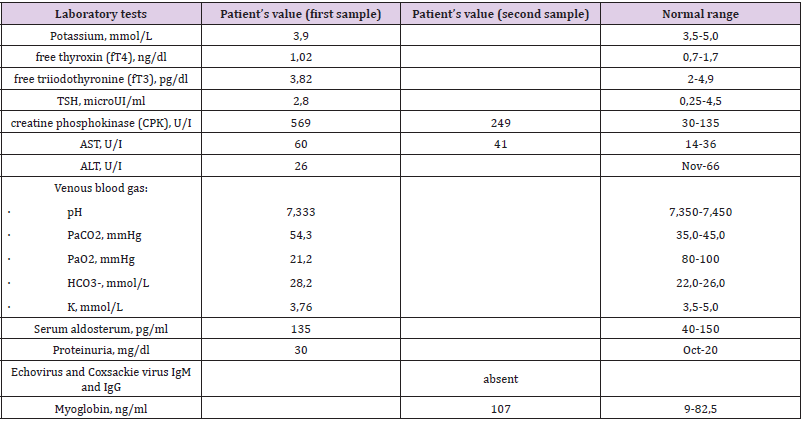
Table 2: Laboratory tests performed and them normal range in relation to our patient’s age.
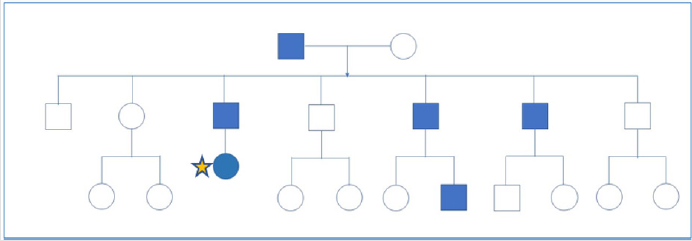
Figure 1: Family pedigree showing three generations. Squares: man; circles: women. Star: our patient. Blue indicates patients with hypokalemic periodic paralysis.
Conclusion
Patient described in this report had a positive family history
for muscle symptoms but there was no established diagnosis in
her family members. Since the presence of poor clinical signs
and normal serum potassium values at admission, we performed
laboratory and instrumental tests to find out possible secondary
causes for the paralytic event [1,16]. A main cause of hypokalemia
is acid-base disorder, so we tested blood gas analysis which showed
compensated acidosis. This finding, associated to normal blood
pressure values, has led us to exclude the diagnostic hypothesis
of Liddle syndrome, postulated in the past for relative [22]. Liddle
syndrome is one of the causes of muscular weakness, especially
in lower limbs, associated with hypokalemia that occurs with
metabolic alkalosis, hypertension, and hypokalemia. Same clinical
manifestations are present in primary hyperaldosteronism,
but aldosterone serum value in our patent was normal. Having
regard to the symptoms’ regression with the assumption of salt
supplements, we concluded that it was a paralytic form due to
hypokalemia [1,16,22]. For this reason, we investigated the major
forms of hypokalemia. Besides Liddle syndrome and primary
hyperaldosteronism, hypokalemia can be found in Gitelman syndrome associated with metabolic alkalosis, hypomagnesemia
and hypocalciuria. In our patient, we did not find any alteration
in urinary and blood electrolytes levels [23-31]. Thyrotoxicosis is
another one of the main causes of secondary periodic hypokalemic
paralysis [32].
Diagnosis is based on laboratory finding of altered thyroid
hormones values (fT3, fT4, TSH); for these parameters there is
no alteration in our patient. Instead, parameters index of muscle
damage like CPK, myoglobin and AST were abnormal. It is
postulated that hypokalemia causes muscle ischemia resulting in
an increase in serum CPK. Elevated CPK levels during the recovery
phase can be used to identify symptomatic patients in whom
serum potassium becomes normal after or during hypokalemic
paralysis. The heterozygous G>A (Arg528His) CACNA1S gene
missense mutation identified in our patient in often triggered
by a high carbohydrate meal, [33] as happened in our case. A
peculiarity of our case is represented by higher levels of CPK, AST
and myoglobin, contrary to what was reported in the series of
Alhasan, et al. [13,33]. Excluding secondary causes and considering
familiar paralytic episodes, genetic analysis in our patient and in
her family was performed [23,24]. Genomic DNA was extracted
from patient’s peripheral blood and from members of her family
using PCR amplification and direct sequencing of CACNA1S exon 11
[5,33].Finally, periodic paralysis in women is mostly asymptomatic
(about 50% of cases) and normokalemic presentation is rarer than
other clinical presentations. Therefore, it is important to have a
clinical suspicion of periodic paralysis, after other causes of muscle
weakness have been ruled out [18].
Consent
Written informed consent was obtained from parents of the patient for the publication of this case report. A copy of the written consent is available for being reviewed by the Editor-in-Chief of this journal.
Competing Interests
The authors declare that they have no competing interests.
References
- Thor MG, Vivekanandam V, Sampedro Castañeda M, Tan SV, Suetterlin K, et al. (2019) Myotonia in a patient with a mutation in an S4 arginine residue associated with hypokalaemic periodic paralysis and a concomitant synonymous CLCN1 mutation. Sci Rep 9(1): 17560.
- Phuyal P, Nagalli S (2021) Hypokalemic Periodic Paralysis. Treasure Island (FL). StatPearls.
- Statland JM, Barohn RJ (2013) Muscle channelopathies: the nondystrophic chas and periodic paralyses. Continuum (Minneap Minn) 19(6 Muscle Disease), pp. 1598-1614.
- Ke T, Gomez CR, Mateus HE, Castano JA, Wang QK (2009) Novel CACNA1S mutation causes autosomal dominant hypokalemic periodic paralysis in a South American family. J Hum Genet 54: 660-664.
- Wang XY, Ren BW, Yong ZH, Xu HY, Fu QX, et al. (2015) Mutation analysis of CACNA1S and SCN4A in patients with hypokalemic periodic paralysis. Mol Med Rep 12: 6267-6274.
- Maggi L, Ravaglia S, Farinato A, Brugnoni R, Altamura C, et al. (2017) Coexistence of CLCN1 and SCN4A mutations in one family suffering from myotonia. Neurogenetics 18: 219-225.
- Matthews E, Portaro S, Ke Q, Sud R, Haworth A, et al. (2011) Acetazolamide efficacy in hypokalemic periodic paralysis and the predictive role of genotype. Neurology 77: 1960-1964.
- Gregor Andelfinger, Andrew R Tapper, Richard C Welch, Carlos G Vanoye, Alfred L George, et al. (2002) KCNJ2 Mutation Results in Andersen Syndrome with Sex-Specific Cardiac and Skeletal Muscle Phenotypes. The American Journal of Human Genetics 71(3): 663-668.
- Morales F, Pusch M (2020) An Up-to-Date Overview of the Complexity of Genotype-Phenotype Relationships in Myotonic Channelopathies. Front Neurol 17(10): 1404.
- Cavel Greant D, Lehmann Horn F, Jurkat Rott K (2012) The impact of permanent muscle weakness on quality of life in periodic paralysis: A survey of 66 patients. Acta Myol 31: 126-133.
- Sansone VA, Ricci C, Montanari M, G Apolone, M Rose, et al. (2012) Measuring quality of life impairment in skeletal muscle channelopathies. Eur J Neurol 19(11): 1470-1476.
- Cannon SC (2015) Channelopathies of Skeletal Muscle Excitability. Compr Physiol 5(2): 761-790.
- Fialho D, Griggs RC, Matthews E (2018) Periodic paralysis. Handb Clin Neurol 148: 505-520.
- Ginanneschi F, Mignarri A, Lucchiari S, Ulzi G, Comi GP, et al. (2017) Neuromuscular excitability changes produced by sustained voluntary contraction and response to mexiletine in myotonia congenita. Neurophysiol Clin 47(3): 247-252.
- Statland JM, Fontaine B, Hanna MG, Johnson NE, Kissel JT, et al. (2018) Review of the Diagnosis and Treatment of Periodic Paralysis. Muscle Nerve 57: 522-530.
- Charles G, Zheng C, Lehmann Horn F, K Jurkat-Rott, J Levitt (2013) Characterization of hyperkalemic periodic paralysis: a survey of genetically diagnosed individuals. J Neurol 260(10): 2606-2613.
- Chalissery AJ, Munteanu T, Langan Y, Brett F, Redmond J (2018) Diverse phenotype of hypokalaemic periodic paralysis within a family. Pract Neurol 18(1): 60-65.
- Andersen ED, Krasilnikoff PA, Overvad H (1971) Intermittent muscular weakness, extrasystoles, and multiple developmental anomalies. Acta Pædiatrica 60: 559-564.
- Basali D, Prayson RA (2015) Episodic weakness, and vacuolar myopathy in hypokalemic periodic paralysis. J Clin Neurosci 22: 1846-1847.
- Sansone V, Meola G, Links TP (2008) Treatment for periodic paralysis. Cochrane Database Syst Rev 2008: CD005045.
- Rolim AL, Lindsey SC, Kunii IS, Fujikawa AM, Soares FA, et al. (2010) Ion channelopathies in endocrinology: Recent genetic findings and pathophysiological insights. Arq Bras Endocrinol Metabol 54(8): 673-681.
- Sansone VA (2019) Episodic Muscle Disorders. Continuum (Minneap Minn) 25(6): 1696-1711.
- Matthews E, Silwal A, Sud R, Michael G Hanna, Adnan Y Manzur, et al. (2017) Skeletal muscle channelopathies: Rare disorders with common pediatric symptoms. J Pediatr 188: 181-185.
- Matthews E, Portaro S, Ke Q, Sud R, Haworth A, et al. (2011) Acetazolamide efficacy in hypokalemic periodic paralysis and the predictive role of genotype. Neurology 77: 1960-1964.
- Conravey A, Santana Gould L (2010) Myotonia congenita and myotonic dystrophy: Surveillance and management. Curr Treat Options Neurol 12: 16-28.
- Jurkat Rott K, Holzherr B, Fauler M (2010) Sodium channelopathies of skeletal muscle result from gain or loss of function. Pflugers Arch 460: 239-248.
- Cannon SC (2010) Voltage-sensor mutations in channelopathies of skeletal muscle. J Physiol 588(pt 11): 1887-1895.
- Johnson NE (2019) Myotonic muscular dystrophies. Continuum (Minneap Minn) 25(6, Muscle and Neuromuscular Junction Disorders), pp. 1682-1695.
- Groome JR, Lehmann Horn F, Fan C, Wolf M, Winston V, et al. (2014) Nav1.4 mutations cause hypokalaemic periodic paralysis by disrupting IIIS4 movement during recovery. Brain 137: 998-1008.
- Bayless Edwards L, Winston V, Lehmann Horn F, Arinze P, Groome JR, et al. (2018) Nav1.4 DI-S4 periodic paralysis mutation R222W enhances inactivation and promotes leak current to attenuate action potentials and depolarize muscle fibers. Sci Rep 8(1): 10372.
- Tella SH, Kommalapati A (2015) Thyrotoxic periodic paralysis: An underdiagnosed and under-recognized condition. Cureus 7: e342.
- Richards S, Aziz N, Bale S, Bick D, Das S, et al. (2015) Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17(5): 405-424.
- Alhasan KA, Abdallah MS, Kari JA, Bashiri FA (2019) Hypokalemic periodic paralysis due to CACNA1S gene mutation. Neurosciences (Riyadh) 24(3): 225-230.
