 Review Article
Review ArticleAbstract
Transcription Factors play a vital role in almost all the biological processes. After the widespread sequencing of genomic and cDNA, it was brought under notice that the legumes encode more than 2,000 transcription factors per genome. Despite of the fact that these transcription factors play pivotal roles in legume revolution, plant development and differentiation, still less than 1% of these factors have been completely characterized genetically. In plants, the transcription factor family containing a Basic Leucine Zipper domain (BZIP domain) is among the chief family of the transcription factors although, these factors are found in all other eukaryotes as well. Here the Genetic Characterization of BZIP transcription factors family will be performed which will ultimately pave a way for the identification and study of the transcription factors of the most complex crop which has been sequenced so far.
Keywords: Soybean; BZIP; Transcription Factors Family; Genetic Characterization
Introduction
Transcription factors are the proteins that bind with DNA and then interact with the other transcriptional regulators including chromatin remodeling or modifying proteins, for blocking or recruiting the access of RNA polymerase to the DNA template. They, play a vital role in almost all the biological processes. The genomes of the plants dedicate approximately 7% of their coding sequences to these vital transcription factors. This justifies the complexity and importance of the transcriptional regulation into the plant genomes. The development and differentiation of plants is chiefly programmed at gene transcription level, which is regulated by transcription factors along with the other proteins which may block or recruit the RNA polymerases access to DNA template [1].
Transcription factors are commonly defined as the sequencespecific DNA-binding proteins which can either repress or activate the transcription process. Now, it has been investigated that the genome of plants encodes more transcription factors than the genome of animals, which is a clear indicator of the fact that the complex nature of transcriptional regulation in plants is at par with that of the animals [2] as nearly every biological process is directly influenced or regulated by these transcription factors [3]. This fact is evident through example that there is a specific class of transcription factors called “general transcription factors” and without these factors, the process of transcription will not occur in eukaryotes [4]. Studies revealed that the transcription factors have been precisely involved in the process of cell development, division, differentiation and migration. The transcription factors of Arabidopsis thaliana have been deeply studied since its genome is being sequenced as a model specie [3,5,6]. After the widespread sequencing of genomic and cDNA, it was brought under notice that the legumes encode more than 2,000 transcription factors per genome. Despite of the fact that these transcription factors play pivotal roles in legume revolution, plant development and differentiation, still less than 1% of these factors have been completely characterized genetically. This pavesa way for the identification and study of the transcription factors of the other newly sequenced crops, like soybean, through comparative analysis and homology searching.
Soybean is known to be a great source of proteins containing the substantial amounts of all the essential amino acids in it. It also contains some of the amino acids that are not even synthesized by the human body (Henkel, 2000). Soybean has now become an important crop in numerous countries around the globe from the past 60 years [7]. Furthermore, Soybean is also known to be the most complex crop which has been sequenced so far. Nowadays, the annotation and identification of Soybean transcription factors has become more convenient after the annotation and prediction of its homology-based genes, which produce the putative protein sequences [8].
In plants, the transcription factor family containing a Basic Leucine Zipper domain (BZIP domain) is among the chief family of the transcription factors although, these factors are found in all other eukaryotes as well. In plants, these transcription factors regulate genes in response to the seed maturation, abiotic stresses, pathogen defense and flower development [3]. This family is present in numerous DNA binding proteins of the eukaryotes. It’s one portion has a region which facilitates the sequence specific binding of DNA and the other portion, called the leucine zipper, which holds the two DNA binding regions together. This DNA binding region consists of numerous basic amino acids like lysine and arginine. Proteins comprising this domain are the transcription factors [9,10]. BZIP transcription factors are found nearly in every eukaryote and BZIP is one of the chief families of the dimerizing transcription factors [11,12]. This paper is concerned with the Genetic Characterization of BZIP transcription factors, which will ultimately pave way for the identification and study of the transcription factors of the most complex crop which has been sequenced so far.
Review of Literature
The use of Bioinformatics approaches has been contributory in identification of the putative Transcription factors in the plants. Transcription factors are commonly defined by using the DNAbinding domain types which are held by the proteins in a family while the putative transcription factor genes are identified mainly due to DNA sequences present inside of the genes which encode the known DNA binding [2,13,14]. Wolfong et al. in 1997 were successful in isolating a cDNA which encodes a novel BZIP protein called G/HBF-1, which is responsible for the binding of the H-box and the adjacent G-box in the proximal section of the chalcone synthase promoter in Soybean.
Haiyang et al. in 2014 proposed through research that the two transcription factors, GmFT2a and GmFT5a, present in soybean, redundantly and differentially control the photoperiod-regulated flowering via the transcriptional up regulation and physical interaction of BZIP’s transcription factor i.e., GmFDl9, which then ultimately catalysis the floral identity genes expression in soybean. Murilo et al. 2013 reviewed that the BZIP transcription factors present in plants are responsive to pathogens also. While Jackoby et al. [2], positioned the BZIP proteins in Arabidopsis into the ten major groups i.e. A, B, C, D, E, F, G, H, I and S. Here, every group of BZIP proteins has a specific sequence which is like the basic region and has common features also, such as the position of the leucine zipper in the protein sequence and the size of the leucine zipper domain.
The classification of BZIP transcription factors (GmbZIPs) in soybean was completed through examination of 47 BZIP sequences along with 75 AtbZIP proteins. Actual outcome was the classification of BZIP transcription factors into10 groups, like was in Arabidopsis. As this classification was done on the basis of the conserved domains. So, it is suitable in plants to for the general classification of BZIP proteins [15]. Heinekamp et al. [16], in 2002 analyzed the tobacco BZIP proteins and found that the BZIP protein BZI-1 exhibits all the distinctive features of a transcription factor. Its function is to bind DNA, particularly the ACGT containing ciselements, and it is localized inside the nucleus of the cell, while its N-terminal domain plays important role as a trans-activation domain in the plant cells.
Liao et al. [15], in 2008 studied 131 BZIP type of transcription factor genes in soybean. They performed the expression analysis of these genes against different stresses, and then from all of the genes, three (03) genes were additionally investigated in relation to stress tolerance, transcriptional activation and DNA-binding specificity. Their results indicated that those transgenic Arabidopsis plants which over express the three additionally investigated genes were less sensitive towards ABA but were more tolerant to the freezing and salt stresses. Soybean uni genes were analyzed by Tian et al. [17] in 2004through EST assembly and as result more than 1,000 transcription factor genes were identified. Arabidopsis possess at least four times as many BZIP genes as worm, yeast and humans [2]. Through molecular and genetic studies of the few of these Arabidopsis thaliana BZIP genes, it is evident that they regulate varied biological processes, for example, stress and light signaling, pathogen defense, flower development and seed maturation. As, BZIP TFs which are contrasting with the functions of WRKY TFs and R2R3-MYB, may contribute to the more diversity being plant-specific if they are early recruited into the plant evolution [18,19]. Chuang et al. [20], in 1999studied the Perianthia genes which are involved in determination of the floral organ number in Arabidopsis thaliana. There, Perianthia genes have homology with the BZIP transcription factors. Moreover, numerous BZIP proteins in plants are known to bind the ACGT cis-acting element, which was identified as the promoters of the plant, bacterial and viral genes [21-26].
Methodology
BZIP transcription factor of soybean was used for the different analysis. MEGA-X, Soybean Knowledge Base databases were used for the performance of different analysis. The Molecular Evolutionary Genetics Analysis or MEGA software is a desktop application which was designed for comparative analysis of homologous gene sequences either from different species or from multi gene families with a superior importance on deducing evolutionary relationships and patterns of protein and DNA evolution. Along with the statistical tools for data analysis, MEGA also contains numerous facilities for convergence of sequenced datasets from web-based repositories or files; moreover, it comprises tools for visual demonstration of the results which are obtained in the interactive form of evolutionary distance matrices and the phylogenetic trees [27]. Soybean Knowledge Base or Soy KB is a comprehensive all-embracing resource of web for translational genomics of Soybean. It was designed for the better integration and management of genomics of soybean, proteomics, metabolomics and transcriptomics data along the annotation of biological pathway and gene function. Moreover, it possesses vital tools like gene family search, Affymetrix probe ID search, protein 3D structure viewer and metabolite search, also the user can upload as well as download annotations and experimental data [28]. The genes of BZIP family downloaded from Soy KB, used for the analysis are shown in Table 1.
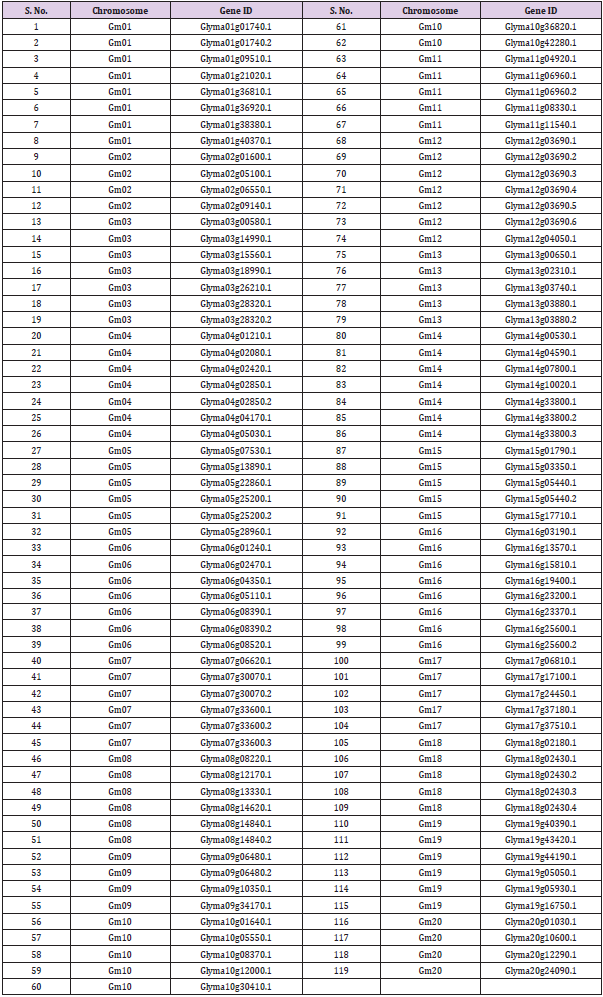
Table 1: Genes of BZIP Family.
Result
Evolutionary Relationships of Taxa
Figure 1 According to Rzhetsky and Nei [29] the history of evolution was derived by using the Minimum Evolution method. Here the sum of branch length of an optimal tree is demonstrated as 175.29000652. Now, like those of the evolutionary distances which are used to infer the phylogenetic tree, similarly a tree is sketched to a scale having branches of the same length units. Moreover, evolutionary distances are computed by using maximum Composite Likelihood method [30] and its units are number of base substitutions per site. The Minimum Evolution tree was examined by utilizing the Close-Neighbor-Interchange (CNI) [31] at a search level of 1. The Neighbor-joining algorithm [32] was utilized for the generation of the initial tree while 119 nucleotide sequences were involved in this analysis while,1st+2nd+3rd+Noncoding were the Codon positions. By using pairwise deletion option all the abstruse positions were discarded for each of the sequence pair. Hence, 1610 positions were in total present in the final dataset while MEGA-X was used for computing the Evolutionary analyses [33].

Figure 1: Evolutionary relationships of taxa.
Use of Maximum Likelihood Method for Evolutionary Analysis
Figure 2 Here, the Maximum Likelihood method and the Tamura-Nei model were used to derive the evolutionary history [34]. The tree carrying the highest log likelihood (-219250.10) is depicted above. Neighbor-Join and Bio NJ algorithms were applied to a matrix of pair wise distances which were estimated by using the Maximum Composite Likelihood (MCL) method to automatically calculate initial tree(s) for heuristic search, and then the selection of topology having superior log likelihood value will be done. The tree is sketched to such scale, in which branch lengths are measured in the number of substitutions per site. Meanwhile, 119 nucleotide sequences were involved in this analysis while1st+2nd+3rd+Noncoding were the Codon positions being included in this analysis. Hence, 1610 positions were in total present in the final dataset while MEGA-X was used for computing the Evolutionary analyses [33].

Figure 2: Use of Maximum Likelihood method for evolutionary analysis.
Evolutionary Relationships in Taxa
Figure 3 According to Saitou and Nei [32] the history of evolution was derived by using Neighbor-Joining method. Here, the sum of branch length of an optimal tree is demonstrated as 200.73212680. Now, like those of the evolutionary distances which are used to infer the phylogenetic tree, similarly a tree is sketched to a scale having branches of the same length units. Moreover, evolutionary distances are computed by using maximum Composite Likelihood method [30] and its units are number of base substitutions per site. So, 119 nucleotide sequences were involved in this analysis while, 1st+2nd+3rd+Noncoding were the Codon positions being included in this analysis. By using pair wise deletion option all the abstruse positions were discarded for each of the sequence pair. Hence, 1610 positions were in total present in the final dataset while MEGA-X was used for computing the Evolutionary analyses [33].

Figure 3: Evolutionary relationships in taxa.
Maximum Parsimony Analysis of Taxa
Figure 4 Here, the Maximum Parsimony method was used to derive the evolutionary history. The evolutionary history was derived by utilizing the Maximum Parsimony method. The most parsimonious tree having length of 80376 is depicted here. There tension index is 0.310626 (0.310626), the consistency index is 0.060193 (0.060193), and the composite index is 0.018697 (0.018697) for all sites and parsimony-informative sites (in parentheses). The MP tree was obtained using the Subtree-Pruning- Regrafting (SPR) algorithm [31] with search level of 0 in which the initial trees were obtained by the random addition of sequences (10 replicates). So, 119 nucleotide sequences were involved in this analysis while, 1st+2nd+3rd+Noncoding were the Codon positions being included in this analysis. Hence, 1610 positions were in total present in the final dataset while MEGA-X was used for computing the Evolutionary analyses [33].
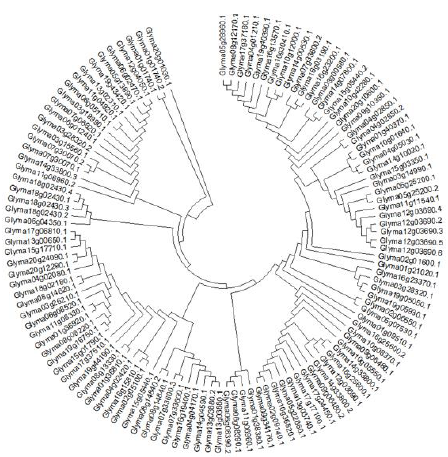
Figure 4: Maximum Parsimony analysis of taxa.
Evolutionary Relationships of Taxa
Figure 5 Here, the UPGMA method was used to derive the evolutionary history [35]. Here, the sum of branch length of an optimal tree is demonstrated as 194. 89363191.Now, like those of the evolutionary distances which are used to infer the phylogenetic tree, similarly a tree is sketched to a scale having branches of the same length units. Moreover, evolutionary distances are computed by using maximum Composite Likelihood method [30] and its units are number of base substitutions per site. So, 119 nucleotide sequences were involved in this analysis while, 1st+2nd+3rd+Noncoding were the Codon positions being included in this analysis. By using pair wise deletion option all the abstruse positions were discarded for each of the sequence pair. Hence, 1610 positions were in total present in the final dataset while MEGA-X was used for computing the Evolutionary analyses [33].

Figure 5: Evolutionary relationships of taxa.
Estimation of Average Evolutionary Divergence over All of the Sequence Pairs
Figure 6 Analysis was conducted by using maximum Composite Likelihood model [30] and the number of base substitutions per site are shown which the average of the overall sequence pairs is. So, 119 nucleotide sequences were involved in this analysis while, 1st+2nd+3rd+Noncoding were the Codon positions being included in this analysis. By using pair wise deletion option all the abstruse positions were discarded for each of the sequence pair. Hence, 1610 positions were in total present in the final dataset while MEGA-X was used for computing the Evolutionary analyses [33].

Figure 6: Estimation of Average Evolutionary Divergence over all of the Sequence Pairs.
Heat Map Analysis
Figure 7A heat map of the 119 BZIP genes is shown here. Individual values contained in a matrix are represented as light and dark colors. Here, red cells denote small values, and red small ones.

Figure 7: Heat Map Analysis.
Conclusion
Transcription factors along with the addition of RNA polymerase complex physically interact along with other proteins to cause variations in the gene transcription [36]. Now, the exact structures of these complexes are still unidentified for most of the plant genes, even though this knowledge is like a precondition to understand the combining control of transcription [37]. Lastly, the genes network being regulated by a “single” transcription factor and its allies are the small part of a greater genetic regulatory network which ensures coordinated expression of genes which are involved in diverse cell related processes throughout from plant differentiation to plant development. So, there is a requirement of incorporation of many of the genomic approaches like functional genomics and bioinformatics to interpret these global gene networks. Hence, the interpretation of these global gene networks would therefore be requiring a combination of the numerous bioinformatic, genomic and functional genomic approaches. A considerable progress in the research of BZIP transcription factor is done over the last 20 years by the application of varied approaches and innovative technologies. Additionally, if BZIPs are considered as the candidate genes in our breeding projects and various other crop improvement programs, it would provide us a vibrant understanding of the several biotic stress-related “signal transduction” events. Hence, it will therefore hint us towards the development of various genetically modified and manipulated crop varieties having the upgraded level of the stress tolerance [38-41].
Conflict of Interests
None.
References
- Udvardi MK, Kakar K, Wandrey M, Montanari O, Murray J, et al. (2007) Legume transcription factors: global regulators of plant development and response to the environment. Plant Physiology 144: 538-549.
- Riechmann JL, Heard J, Martin G, Reuber L, Jiang CZ, et al. (2000) Arabidopsis transcription factors: genome-wide comparative analysis among eukaryotes. Science 290: 2105-2110.
- Jakoby M, Weisshaar B, W Dröge-Laser, Vicente-Carbajosa J, Tiedemann J, et al. (2002) bZIP transcription factors in Arabidopsis. Trends in plant science 7: 106-111.
- Weinzierl RO (2008) Mechanisms of gene expression: structure, function and evolution of the basal transcriptional machinery.
- Liu Q, Kasuga, M, Sakuma Y, Abe H, Miura S, et al. (1998) Two transcription factors, DREB1 and DREB2, with an EREBP/AP2 DNA binding domain separate two cellular signal transduction pathways in drought-and low-temperature-responsive gene expression, respectively, in Arabidopsis. The Plant Cell 10: 1391-1406.
- Ülker B, Somssich (2004) WRKY transcription factors: from DNA binding towards biological function. Current opinion in plant biology 7: 491-498.
- Carpenter JE, Gianessi LP (2001) Agricultural biotechnology: Updated benefit estimates (pp. 1-46). Washington, DC: National Center for Food and Agricultural Policy.
- Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, et al. (2010) Genome sequence of the palaeopolyploid soybean. Nature 463: 178.
- Ellenberger T (1994) Getting a grip on DNA recognition: structures of the basic region leucine zipper, and the basic region helix-loop-helix DNA-binding domains. Current Opinion in Structural Biology 4: 12-21.
- Hurst HC (1995) Transcription factors 1: bZIP proteins. Protein profile 2: 101-168.
- Amoutzias GD, Robertson DL, Van de Peer Y, Oliver SG (2008) Choose your partners: dimerization in eukaryotic transcription factors. Trends in biochemical sciences 33: 220-229.
- Amoutzias GD, Veron AS, Weiner III J, Robinson-Rechavi M, Bornberg-Bauer E, et al. (2006) One billion years of bZIP transcription factor evolution: conservation and change in dimerization and DNA-binding site specificity. Molecular biology and evolution 24: 827-835.
- Iida K, Seki M, Sakurai T, Satou M, Akiyamac K, et al. (2005) RARTF: database and tools for complete sets of Arabidopsis transcription factors. DNA Research 12: 247-256.
- Guo A, He K, Liu D, Bai S, Gu X, et al. (2005) DATF: a database of Arabidopsis transcription factors. Bioinformatics 21: 2568-2569.
- Liao Y, Zou HF, Wei W, Hao YJ, Tian AG, et al. (2008) Soybean GmbZIP44, GmbZIP62 and GmbZIP78 genes function as negative regulator of ABA signaling and confer salt and freezing tolerance in transgenic Arabidopsis. Planta 228: 225-240.
- Heinekamp T, Kuhlmann M, Lenk A, Strathmann A, Dröge-Laser W (2002) The tobacco bZIP transcription factor BZI-1 binds to G-box elements in the promoters of phenylpropanoid pathway genes in vitro, but it is not involved in their regulation in vivo. Molecular Genetics and Genomics 267: 16-26.
- Tian AG, Wang J, Cui P, Han YJ, Xu H, et al. (2004) Characterization of soybean genomic features by analysis of its expressed sequence tags. Theoretical and Applied Genetics 108: 903-913.
- Eulgem T, Rushton PJ, Robatzek S, Somssich IE (2000) The WRKY superfamily of plant transcription factors. Trends in plant science 5: 199-206.
- Stracke R, Werber M, Weisshaar B (2001) The R2R3-MYB gene family in Arabidopsis thaliana. Current opinion in plant biology 4: 447-456.
- Chuang CF, Running M, Williams R,Meyerowitz EM (1999) The PERIANTHIA gene encodes a bZIP protein involved in the determination of floral organ number in Arabidopsis thaliana. Genes & development 13: 334-344.
- Mikami K, Tabata T, Kawata T, Nakayama T, Iwabuchi M (1987) Nuclear protein (s) binding to the conserved DNA hexameric sequence postulated to regulate transcription of wheat histone genes. FEBS letters 223: 273-278.
- Giuliano G, Pichersky E, Malik VS, Timko MP, Scolnik PA, et al. (1988) An evolutionarily conserved protein binding sequence upstream of a plant light-regulated gene. Proceedings of the National Academy of Sciences 85: 7089-7093.
- Lam E, Benfey PN, Gilmartin PM, Fang RX, Chua NH (1989) Site-specific mutations alter in vitro factor binding and change promoter expression pattern in transgenic plants. Proceedings of the National Academy of Sciences 86: 7890-7894.
- Bouchez D, Tokuhisa JG, Llewellyn DJ, Dennis ES, Ellis JG (1989) The ocs‐element is a component of the promoters of several T‐DNA and plant viral genes. The EMBO journal 8: 4197-4204.
- Schindler U, Beckmann H, Cashmore AR (1992) TGA1 and G-box binding factors: two distinct classes of Arabidopsis leucine zipper proteins compete for the G-box-like element TGACGTGG. The Plant Cell 4: 1309-1319.
- Schindler U, Menkens AE, Beckmann H, Ecker JR, Cashmore AR (1992) Heterodimerization between light‐regulated and ubiquitously expressed Arabidopsis GBF bZIP proteins. The EMBO journal 11: 1261-1273.
- Kumar S, Nei M, Dudley J, Tamura K (2008) MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Briefings in bioinformatics 9: 299-306.
- Joshi T, Patil K, Fitzpatrick MR, Franklin LD, Yao Q, et al. (2012) Soybean Knowledge Base (SoyKB): a web resource for soybean translational genomics. In BMC genomics 13(1): S15.
- Rzhetsky A, Nei M (1992) A simple method for estimating and testing minimum-evolution trees.
- Tamura K, Nei M, Kumar S (2004) Prospects for inferring very large phylogenies by using the neighbor-joining method. Proceedings of the National Academy of Sciences 101: 11030-11035.
- Nei M, Kumar S (2000) Molecular Evolution and Phylogenetics. Oxford University Press, New York, USA.
- Saitou, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Molecular biology and evolution 4: 406-425.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Molecular biology and evolution 35: 1547-1549.
- Tamura K, Nei M (1993) Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Molecular biology and evolution 10: 512-526.
- Sneath PH, Sokal RR (1973) Numerical taxonomy. The principles and practice of numerical classification.
- Lee TI, Young RA (2000) Transcription of eukaryotic protein-coding genes. Annual review of genetics 34: 77-137.
- Singh KB (1998.) Transcriptional regulation in plants: the importance of combinatorial control. Plant Physiology 118: 1111-1120.
- Alves M, Dadalto S, Gonçalves A, De Souza G, Barros V, et al. (2013) Plant bZIP transcription factors responsive to pathogens: a review. International J of molecular Sci 14: 7815-7828.
- Dröge‐Laser W, Kaiser A, Lindsay WP, Halkier BA, Loake GJ, et al. (1997) Rapid stimulation of a soybean protein‐serine kinase that phosphorylates a novel bZIP DNA‐binding protein, G/HBF‐1, during the induction of early transcription‐dependent defenses. The EMBO Journal 16: 726-738.
- Henkel J (2000) Soy. Health claims for soy protein, questions about other components. FDA consumer 34: 13-15.
- Nan H, Cao D, Zhang D, Li Y, Lu S, et al. (2014) GmFT2a and GmFT5a redundantly and differentially regulate flowering through interaction with and upregulation of the bZIP transcription factor GmFDL19 in soybean. PloS one 9: e97669.
