 Research Article
Research ArticleAbstract
Purpose: Our aims were to study the incidence, age at diagnosis, familial aggregation of cancer, clinical presentation, delay/pitfalls in the diagnosis and overall survival of the different types of breast angiosarcomas
Material and Methods: We investigated all breast angiosarcomas diagnosed in the area of Vigo (Galicia, Spain) during the period between January 1974 and December 2020.
Results: Twelve patients (11 women and 1 man) with breast angiosarcoma were identified among a total of 5000 malignant neoplasms of the breast: 3 primary angiosarcomas and 9 secondary angiosarcomas (7 post-radiotherapy and 2 Stewart- Treves syndromes - associated with lymphedema). Globally, the 12 breast angiosarcomas represent 0.24% of all malignant breast cancers (BCs) in our healthcare area; that is to say, one breast angiosarcoma is diagnosed every 400 BCs. The mean age at diagnosis was 66.5 years (range: 28-91). We highlight the intense familial aggregation of cancer (present in almost all cases) but genetic studies have only been performed in two of them. All the primary angiosarcomas appeared as palpable nodules, while the secondary ones showed a more heterogeneous clinical picture (cutaneous oedema, hematomas, red-bluish macules or even as nodules). This non-specific clinical presentation caused errors and was one of the causes of the diagnostic delay. The mean interval between the diagnosis of BC and angiosarcoma was 106 months (SD 46). Angiosarcomas showed a very aggressive behaviour: 8 of the 12 patients died, and survival ranged from 3 months to 19 months after diagnosis, with high rates of local and distant progression.
Conclusions: Breast angiosarcoma is a neoplasm with a very poor prognosis. Due to its rarity and its different forms of presentation, it represents a diagnostic challenge. We have observed an intense and striking familial aggregation of cancer that leads us to think that there is a genetic predisposition that plays an important role in the carcinogenesis of breast angiosarcomas. Investigating and clarifying this predisposition could have consequences in its prevention and in the treatment planning (eg radiotherapy) of patients with BC.
Keywords: Breast Sarcoma; Breast Angiosarcoma; Primary Angiosarcoma; Radiation-Induced Angiosarcoma; Stewart- Treves Syndrome; Breast Cancer
Introduction
Breast sarcomas are histologically heterogeneous non-epithelial malignant tumours derived from mesenchymal cells, arising from mammary connective tissue. They are rare, representing less than 1% of all breast cancers (BCs) and less than 5% of all soft tissue sarcomas [1,2]. The subtype of sarcoma strongly linked to the breast is the angiosarcoma, originating in the endothelial vascular cells. It is a very aggressive tumor, with rapid proliferation and infiltration of the surrounding tissues; generally associated with a poor prognosis [3]. Angiosarcomas can be “primary” when they originate de novo or “secondary” to BC treatment (secondary to radiotherapy or in the context of arm/breast/chest wall lymphedema). Therefore, we have three different clinicopathological entities or subtypes of angiosarcoma related to the breast: primary angiosarcoma of the breast [4], radiation-induced angiosarcoma [5] and angiosarcoma that arises in areas with chronic lymphedema (also called Stewart- Treves syndrome) [6]. In this article, we present the series of 12 cases of breast angiosarcomas diagnosed in our healthcare area in the last decades and discuss the frequency, aetiology, clinical presentation, age at diagnosis, familial aggregation of cancer, diagnostic delay and outcome of this rare breast neoplasm.
Material and Methods
All cases of breast angiosarcomas diagnosed in different hospitals in the health area of Vigo (Municipal Hospital of Vigo, Xeral University Hospital of Vigo, Meixoeiro Hospital, Álvaro Cunqueiro University Hospital and Povisa Hospital) during the period between January 1974 and December 2020 were included in the study. The medical records of the 12 cases were studied retrospectively and prospectively. Personal data, familial aggregation of cancer and clinical presentation were included; as well as radiological, histological and immunohistochemical findings. An individualized clinical follow-up of all patients allowed us to know the exact moment of local recurrences and distant metastases, as well as the date and cause of death. The data obtained in this study were entered into a computer database developed in the Microsoft Excel program. The statistical analysis was carried out with the statistical program SPSS-PC for Windows.
Results
Of the total of 5000 BCs in our series, a total of 12 malignant neoplasms of the breast were classified as breast angiosarcomas: 3 primary angiosarcomas and 9 secondary angiosarcomas (7 radiation-induced angiosarcoma and 2 Stewart-Treves Syndromes). Breast angiosarcomas represent 0.24% of all breast malignant neoplasms in our series. Therefore, around one breast angiosarcoma is diagnosed for every 400 BCs (Table 1). The mean age at the time of diagnosis of breast angiosarcomas was 66.5 years (range: 28 - 91 years). No statistically significant differences were found between the age of presentation and the types of angiosarcoma (Table 2). The mean interval between the diagnosis of BC and secondary angiosarcoma was 106.1 months (SD 46). The interval ranged from 3 years and 11 months (the shortest) to 12 years and 10 months (the longest) (Tables 3 & 4). There were no statistically significant differences in the diagnostic interval time between the groups of secondary angiosarcomas (post-radiotherapy vs. Stewart-Treves syndrome) (p >0.05) (Table 3). Noteworthy is that the three cases of primary angiosarcoma presented as a “palpable nodule”. The age of the youngest patient (28 years) led to a significant diagnostic delay since the ultrasound imaging findings suggested benignity. In this case, the rapid growth of the tumor led to a more interventional aptitude, which provided the diagnosis. The other two women with primary breast angiosarcomas, aged 56 and 66, had a history of reduction mammoplasty and previous trauma, respectively (Table 4). The clinical presentation of the two women with angiosarcomas secondary to chronic lymphedema (Stewart-Treves syndrome) was in the form of bluish-red macules on the skin of the arm with lymphedema. Angiosarcomas secondary to radiotherapy showed a more heterogeneous clinical picture with retraction or skin induration in some cases, in the form of a hematoma in another, or as a bluish-red macula or as a palpable nodule. The only man in our series of secondary angiosarcomas showed a palpable nodule at the level of the irradiated upper limb (arm) showing small lymphedema (Table 4).
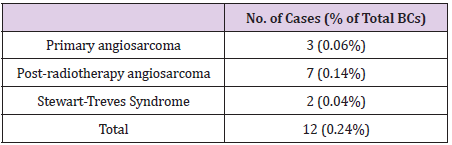
Table 1: Frequency of breast angiosarcoma in relation to the total series of BC.
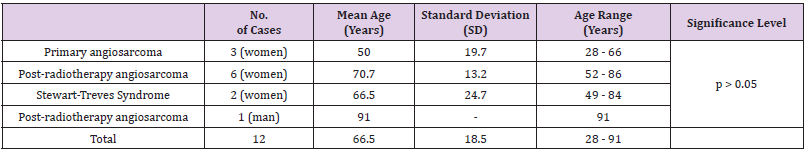
Table 2: Age at the time of diagnosis of breast angiosarcoma.
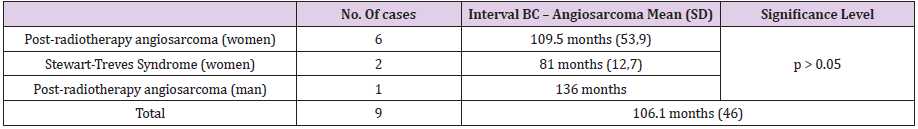
Table 3: Interval between the diagnosis of BC and secondary breast angiosarcoma.
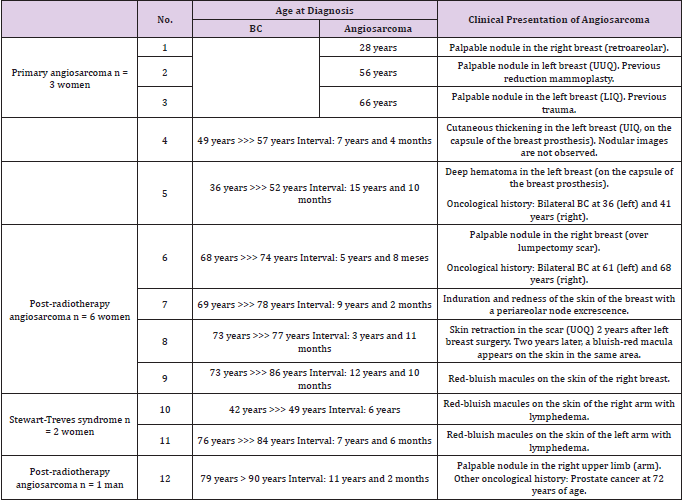
Table 4: Ages and intervals of presentation of BC and angiosarcoma & clinical presentation of breast angiosarcoma.
Note: UUQ (union of upper quadrants), LIQ (low inner quadrant), UIQ (upper inner quadrant), UOQ (upper outher quadrant).
obstetric history and the menarche did not show any
significant findings, apart from the fact that 2 of the women with
primary angiosarcomas were nulliparous and the other patient with
primary angiosarcoma had only a single child at the age of 23 years.
On the contrary, the women with secondary angiosarcomas had a
greater offspring (Table 5). However, what caught our attention is
the intense familial aggregation of cancer (Table 5). Unfortunately,
so far genetic studies have only been started in two of the twelve
cases: in one of them the panel of genes associated with BC was
negative (case number 12); and in the other patient (case number
2), two point mutations were found (in the CHEK2 gene (c.349A>
G, p. Arg117Gly) and in the PALB2 gene (exon4: c.1010T> C, p.
Leu337Ser)). Due to the fact that it is a rare disease and due to its
heterogeneous presentation, we frequently observe delays and/or
errors in diagnosis. We detail them individually in Table 6.
50% of breast angiosarcomas (6 of 12) were classified,
according to histological grade, as high histological grade: poorly
differentiated angiosarcomas. Overall, their behaviour was very
aggressive; causing death in 8 of the 12 cases in our series, with a
survival range that ranged from 3 to 19 months; with high rates of
local and remote progression. One of the women with angiosarcoma
died of metastasis from the previous BC. The follow-up time for
female survivors is too short to draw definitive conclusions (Table
7).
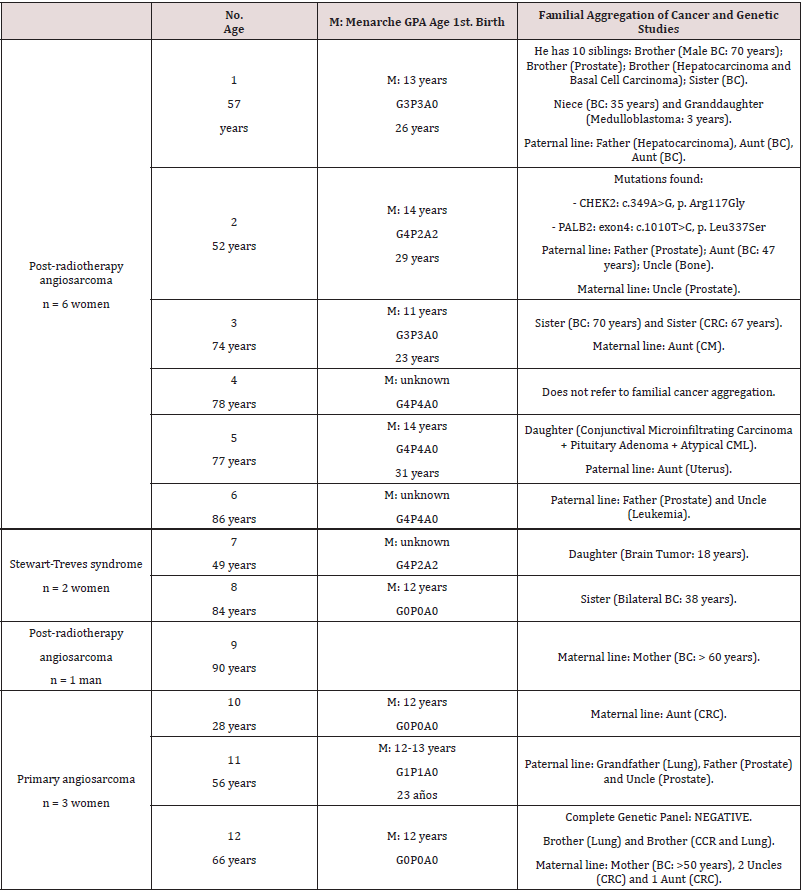
Table 5: Angiosarcoma: Obstetric history and familial aggregation of cancer (genetic studies).
Note: G = Gravida (Number of Pregnancies), P = Para (Number of Births of Viable Offspring), A = Abortus, CRC (Colo-Rectal Carcinoma), CML (Chronic Myeloid Leukemia).
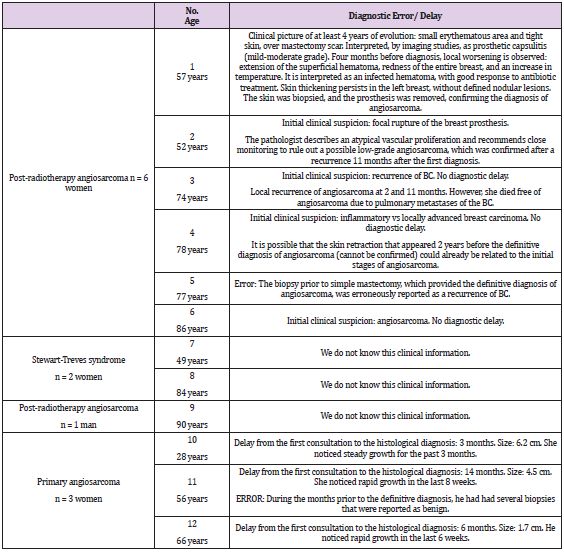
Table 6: Delay and/or errors in the diagnosis of breast angiosarcoma.
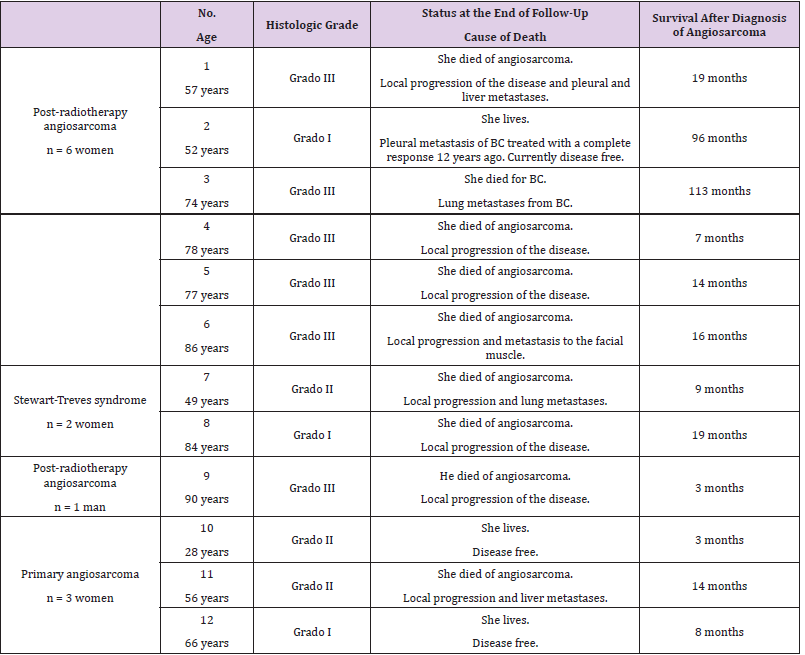
Table 7: Breast angiosarcoma: histological grade, survival (months) and cause of death.
Discussion
Breast angiosarcoma is a rare clinical entity. It arises from the mammary stroma and is the most common sarcomatous malignancy in the mammary gland. It originates from endothelial cells that line blood (and/or lymphatic) vessels. We distinguish three different clinical entities: primary angiosarcomas and two different types of secondary angiosarcomas (those that arise after radiotherapy for a previous BC and those that arise on an area - breast or arm - with intense chronic lymphedema after surgical treatment of a BC with or without radiation therapy, also called Stewart-Treves syndrome). Due to its extreme rarity, the specific incidence is difficult to define. In our series, only 12 angiosarcomas were identified among a series of 5000 malignant neoplasms of the breast, which represented 0.24%. This means that at least in our environment, a breast angiosarcoma will be diagnosed approximately for every 400 BCs. Wang XY, et al. [7] in a similar series of 5000 BC identified 11 angiosarcomas, which reinforces our findings regarding the incidence of this entity. As in our series, these authors only found three patients with primary breast angiosarcomas. Therefore, we conclude that the incidence of primary angiosarcoma is 0.06% of all BCs. Secondary angiosarcomas, although more frequent than primary ones, are also a very rare clinical entity: 0.18% of all BCs in our series (0.14% in the case of post-radiotherapy angiosarcomas and only 0.04% Stewart-Treves syndromes). Primary angiosarcomas tend to occur at younger ages, often in women younger than 40 years (between 20 and 40 years) [8,9]. However, in our series the three women diagnosed with primary angiosarcomas were 28, 56 and 66 years old, respectively (mean age: 50, range: 28-66). The mean age at the time of diagnosis of secondary angiosarcomas is higher than the age of primary angiosarcomas (median 64, range: 44-84 years) [10]. In our series, the mean age of post-radiotherapy angiosarcomas was 70.7 years (range: 52-86) and 66.5 years for Stewart-Treves syndrome (range: 49-84 years). The only man in the series with secondary breast angiosarcoma was diagnosed at age 91.
Secondary radiation-induced angiosarcomas follow the criteria established by Cahan, et al. in 1948 [11], which were modified by Arlen et al. in 1971 [12]: a location in the previous field of radiotherapy, a latency period of at least 3 years (period ranging from 1 to 26 years in the literature) (Table 8) and a histological discordance between primary and secondary tumor. Secondary angiosarcoma associated with chronic lymphedema was first described in 1948 by Stewart and Treves, since then it has also been known as Stewart-Treves syndrome. It arises after a period ranging from 4 to 27 years and develops on the limb with lymphedema or on the chest wall itself after a mastectomy with axillary lymphadenectomy [13]. In most of these secondary cases there is evidence of tissue injury induced by both radiation and lymphedema. One of the long-term complications of radiotherapy is the development of secondary malignancies, which was a debate when breast conserving treatment for BC emerged (showing that lumpectomy plus radiotherapy obtained similar results to mastectomy plus lymphadenectomy in terms of survival) [14-23]. Huang and Mackillop [24] demonstrated that BC patients treated with radiotherapy had a 15.9-fold increased risk of developing angiosarcomas compared to controls. However, we will always have to take into account the extreme rarity of this clinical entity. Radiation therapy can increase the risk by causing radiationinduced mutations in exposed tissues (mammary gland, chest wall and upper limb) and also by contributing to the development of lymphedema. It is obvious that the risk of developing an angiosarcoma does not outweigh the benefit of current treatment protocols. Therefore, radiotherapy continues to play a critical role in the treatment of BC [25]. Furthermore, technological advances have significantly improved the field of radiation therapy exposure. Also, the almost generalized implementation of selective sentinel node biopsy has decreased the incidence of lymphedema, helping to reduce the risk of radiotherapy-induced carcinogenesis. A recent population-based study estimated the risk of radiationinduced angiosarcoma to be approximately 0.1% [22]. Primary angiosarcoma usually presents as a painless, ill-defined palpable mass that grows rapidly; reaching a size greater than 4 cm in the majority of reported cases (2/3 cases in our series exceeded 4 cm) [26, 27]. In our series, the three primary angiosarcomas presented as a “palpable nodule”. Frequently, the rarity of this entity and sometimes the extremely young age of these women cause errors and excessive delays in diagnosis (Table 6). Our youngest patient (28 years old) had consulted months before for a breast lump whose ultrasound imaging findings suggested benignity. Finally, the rapid growth rate of the tumor required a more interventional aptitude, which provided the definitive diagnosis.
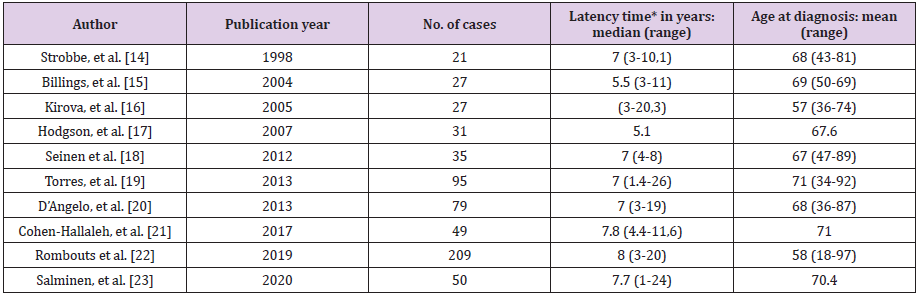
Table 8: Literature review: Clinical studies that include secondary radiation-induced angiosarcomas (latency time and age at diagnosis).
Note: *Latency time: time interval between radiation therapy and diagnosis of angiosarcoma.
However, the clinical presentation of secondary angiosarcomas is more varied. Its heterogeneous presentation and its rarity contribute to a significant delay between the first symptom and the definitive diagnosis [26-27]. Secondary angiosarcomas can present in the form of palpable nodules, as a hematoma or with essentially cutaneous symptoms in the form of macules, plaques or nodules with a red-purplish coloration that can simulate benign skin lesions. If they are not diagnosed and treated, they progress and increase in size, and may be complicated by ulcers or edema [28]. The growth is explosive in high-grade tumours and more insidious in low-grade ones. In our series, the clinical presentation coincides with that described in the literature: palpable nodule for primary angiosarcomas and more heterogeneous skin lesions in the case of secondary angiosarcomas. One of the reasons that contributes to the diagnostic delay is explained by the fact that imaging findings can be nonspecific and inconclusive [29]. Mammography may reveal a thickening of the skin and an ill-defined superficial mass. Ultrasound usually shows a heterogeneous hyper vascular mass associated with architectural distortion. These findings are nonspecific, especially if they are located near the dermis, and may suggest benign skin changes induced by radiation therapy. Magnetic resonance imaging is more reliable, it allows determining the extent of the lesion, and it allows recognizing a heterogeneous mass with lakes of blood that increase rapidly due to haemorrhages. Angiosarcomas usually metastasize hematogenous; making lymphadenectomy unnecessary since the lymph nodes in the armpit are usually not affected.
The definitive diagnosis is provided by biopsy and histological study, being necessary to use immunohistochemical techniques that confirm the endothelial nature of the tumor (antigen related to factor VIII, vascular endothelial growth factor - VEGF -, CD34 and CD31 - the most common and with the highest sensitivity and specificity for angiosarcomas -) [25]. Most of them are poorly differentiated tumours, a fact that also coincides with the findings of our series (grade III in 50%). A source of error for the pathologist is low-grade angiosarcomas that can be erroneously interpreted as benign lesions, especially when the sample is small, and the clinician does not suggest to the pathologist the possibility of angiosarcoma as a differential diagnosis. In our series, we have observed a striking and intense familial aggregation of cancer. This fact has not been widely studied in the literature [30] and perhaps we should begin to take it more into account, since it could allow us to select a group of patients with a higher risk of developing radiation-induced angiosarcomas and consider avoiding this type of treatment, as far as possible. Most of the genes that predispose to cancer are involved in DNA repair. Therefore, it is plausible that those patients with certain genetic alterations related to different Hereditary Cancer Syndromes were more susceptible to developing angiosarcomas as well.
Bree, et al. in 2002 [31] demonstrated that genetic predisposition plays a fundamental role in the development of secondary angiosarcomas. More recently, Kadouri, et al. [32] found a high frequency of BRCA1 and BRCA2 mutations in patients diagnosed with post-radiotherapy angiosarcoma and estimated the risk for carriers to be approximately twice as high. Jiang W et al. describe a case of angiosarcoma in a BRCA1 positive patient (187delAG) with a previous bilateral BC [33]. A high percentage of p53 mutations associated with angiosarcoma cases was also found [34,35]. Li-Fraumeni Syndrome is an autosomal dominant inherited disorder that presents a greater susceptibility to radiation; therefore, the indication for radiation therapy in these patients is controversial [36,37]. The investigations carried out by the group of Thibodeau BJ el al. demonstrate a mutational pattern associated with genes involved in DNA repair [38]. In our series, the intense familial aggregation is striking, but only two patients underwent a genetic study (Table 5). At present, advances in clinical and molecular genetics and the possibility of breast conserving therapy (which involves radiotherapy) in a large part of patients with BC force us to ask ourselves and investigate if we can avoid or reduce the appearance of secondary angiosarcomas. It is necessary to promote good genetic counselling. The simplest and cheapest tool is the elaboration of a correct family tree that has to be updated as time goes by. We must not forget that sometimes cancers in other members of the family are subsequent to the diagnosis of BC, hence familial aggregation of cancer is more important than simple familial history of cancer.
The low incidence of secondary angiosarcomas in BC patients makes this investigation difficult. There is currently no clear consensus on its management. Surgical resection with negative margins is the mainstay of treatment, either by conservative surgery or mastectomy for breast lesions [9,39]. In the case of involvement of the upper extremities, a wide local excision or even an amputation can be done. Surgical excision with wide margins is associated with a better prognosis [23]. Elective axillary lymphadenectomy is not justified, given the low frequency of regional lymph node metastases (11-20%); it is only performed if the armpit is clinically positive. The choice of adjuvant therapy is even more debatable. Adjuvant radiotherapy appears to have an additional benefit, especially in high-grade tumors or large neoplasms, as well as when the margins are positive or very close [40]. However, the role of adjuvant chemotherapy remains questionable; except in high-grade tumors (where it seems more beneficial) and in locally advanced cases where it becomes the mainstay of treatment [40]. Angiosarcoma is an aggressive, easily infiltrative tumor with a high rate of local recurrence and metastasis [15,41]. A fact that was also observed in our series: locoregional progression/recurrence rate of 66.7% and metastasis rate of 33.3%. The most significant prognostic factor for recurrence is the size of the lesion at diagnosis. Therefore, an early diagnosis is essential if we want to increase survival rates [41]. Local recurrences are common (45-60%) despite extensive tumor resection. Reported rates of advanced/metastatic disease at presentation range from 16% to 44%, and overall survival ranges from 6 to 16 months [42]. In our series, angiosarcoma caused death in 8 of the 12 cases, with a survival interval in these cases that ranged from 3 to 19 months.
Conclusion
Breast angiosarcoma is a very rare clinical entity with a very poor prognosis due to its high rates of local and distant recurrence. The young age at the time of diagnosis of most primary angiosarcomas and the low incidence of both primary and secondary angiosarcomas together with the difficulties in their diagnosis cause diagnostic delays that condition their survival. Its diagnosis continues to be a challenge due to its non-specificity from the clinical, radiological and histological point of view. The intense familial aggregation of cancer in these families may be reflecting a genetic predisposition so far little studied. Advances in clinical genetics, molecular genetics and sequencing techniques should allow us to investigate and deepen the genetic susceptibility of breast angiosarcomas. It is important to know this genetic susceptibility in order to be able to select those patients with a higher risk of radiation-induced carcinogenesis and thus consider substituting radiotherapy for other alternative therapies with similar survival rates, as is the case of mastectomy versus more conservative approaches in selected cases.
References
- May DS, Stroup NE (1991) The incidence of sarcomas of the breast among women in the United States, 1973-1986. PlastReconstrSurg 87(1): 193-194.
- Mc Gowan TS, Cummings BJ, O'Sullivan B, Catton CN, Miller N, et al. (2000) Ananalysis of 78 breast sarcoma patientswithoutdistantmetastases at presentation. Int J RadiatOncolBiolPhys 46(2): 383-390.
- Gutkin PM, Ganjoo KN, Lohman M, Von Eyben R, Charville GW, et al. (2020) Angiosarcoma of theBreast: Management and Outcomes. Am J ClinOncol 43(11): 820-825.
- Rodríguez Fernández V, Cameselle Cortizo L, García Mallo A, González L, Marín E, et al. Primary Breast Angiosarcoma: A Rare and Challenging Case Report. Archives of Oncology and Cancer Therapy 3(1): 12-25.
- Rodríguez Fernández V, Cameselle Cortizo L, Novo Domínguez A, Villar Fernández B, De Castro Parga G, et al. (2020) Angiosarcoma after radio therapy for male breast cancer: A rare clinical case. Cancer Therapy 3: 1-4.
- Rodríguez Fernández V, Cameselle Cortizo L, Valdés Pons J, Novo A, Figueiredo Alonso E, et al. (2020) Stewart-Treves Syndrome Involving Chronic Lymphedema Postmastectomy: a Case Report and Review of the Literature. International Journal of Medical Science and Health Research 4(5): 85-92.
- Wang XY, Jakowski J, Tawfik OW, Thomas PA, Fan F (2009) Angiosarcoma of the breast: a clinic pathologic analysis of cases from the last 10 years. Ann Diagn Pathol 13(3): 147-150.
- Shet T, Malaviya A, Nadkarni M, Kakade A, Parmar V, et al. (2006) Primary angiosarcoma of the breast: observations in Asian Indian women. J Surg Oncol 94(5): 368-374.
- Abdou Y, Elkhanany A, Attwood K, Ji W, Takabe K, et al. (2019) Primary and secondary breast angiosarcoma: single center report and a meta-analysis. Breast Cancer Res Treat 178(3): 523-533.
- Vorburger SA, Xing Y, Hunt KK, Lakin GE, Benjamin RS, et al. (2005) Angiosarcoma of the breast. Cancer 104(12): 2682-2688.
- Cahan WG, Woodard HQ, Higinbotham NL, Stewart FW, Coley BL (1998) Sarcoma arising in irradiatedbone: report of eleven cases. Cancer 82(1): 8-34.
- Arlen M, Higinbotham NL, Huvos AG, Marcove RC, Miller T, et al. (1971) Radiation-induced sarcoma of bone. Cancer 28(5): 1087-1099.
- Kaklamanos IG, Birbas K, Syrigos KN, Vlachodimitropoulus D, Goutas N, et al. (2011) Breast angiosarcoma that is no trelated to radiation exposure: a comprehensive review of the literature. Surg Today 41(2): 163-168.
- Strobbe LJ, Peterse HL, Van Tinteren H, Wijnmaalen A, Rutgers EJ (1998) Angiosarcoma of the breast after conservation therapy for invasive cancer, the incidence and outcome. Anun for seen sequela. Breast Cancer Res Treat 47(2): 101-109.
- Billings SD, Mc Kenney JK, Folpe AL, Hardacre MC, Weiss SW (2004) Cutaneous angiosarcoma following breast-conserving surgery and radiation. An analysis of 27 cases. Am J Surg Pathol 28(6): 781-788.
- Kirova YM, Vilcoq JR, Asselain B, Sastre Garau X, Fourquet A (2005) Radiation-induced sarcomas after radio therapy for breast carcinoma: a large-scale single-institution review. Cancer 104(4): 856-863.
- Hodgson NC, Bowen Wells C, Moffat F, Franceschi D, Avisar E (2007) Angiosarcomas of the breast. A review of 70 cases. Am J Clin Oncol 30(6): 570-573.
- Seinen JM, Styring E, Verstappen V, Vult von Steyern F, Rydholm A, et al. (2012) Radiation-associated angiosarcoma after breast cancer: high recurrencerate and poor survival despite surgical treatment with R0 resection. Ann Surg Oncol 19(8): 2700-2706.
- Torres KE, Ravi V, Kin K, Yi M, Guadagnolo BA, et al. (2013) Long-term outcomes in patients with radiation-associated angiosarcomas of the breast following surgery and radio therapy for breast cancer. Ann SurgOncol 20(4): 1267-1274.
- D’Angelo SP, Antonescu CR, Kuk D, Qin L, Moraco N, et al. (2013) High-risk features in radiation-associated breast angiosarcomas. Br J Cancer 109(9): 2340-2346.
- Cohen Hallaleh RB, Smith HG, Smith RC, Stamp GF, Al Muderis O, et al. (2017) Radiation induced angiosarcoma of the breast: outcomes from a retrospective case series. Clin Sarcoma Res 7: 15.
- Rombouts AJM, Huising J, Hugen N, Siesling S, Poortmans PM, et al. (2019) Assessment of Radiotherapy-Associated Angiosarcoma After Breast Cancer Treatment in a Dutch Population-Based Study. JAMA Oncol 5(2): 267-269.
- Salminen SH, Wiklund T, Sampo MM, Tarkkanen M, Pulliainen L, et al. (2020) Treatment and Prognosis of Radiation-Associated Breast Angiosarcoma in a Nationwide Population. Ann Surg Oncol 27(4): 1002-1010.
- Huang J, Mackillop WJ (2001) Increased risk of soft tissue sarcoma after radio therapy in women with breast carcinoma. Cancer 92(1): 172-180.
- Shah S, Rosa M (2016) Radiation-associated angiosarcoma of the breast: clinical and pathologic features. Arch Pathol Lab Med 140(5): 477-481.
- Bordoni D, Bolletta E, Falco G, Cadenelli P, Rocco N, et al. (2016) Primary angiosarcoma of thebreast. Int J Surg Case Rep 20S: 12-15.
- Georgiannos SN, Sheaff M (2003) Angiosarcoma of the breast: a 30 year perspective with an optimistic outlook. British Journal of Plastic Surgery 56(2): 129-134.
- Zemanova M, Machalekova K, Sandorova M, Boljesikova E, Skultetyova M, et al. (2013) Clinical management of secondary angiosarcoma after breast conservation therapy. Rep Pract Oncol Radiother 19(1): 37-46.
- Sheppard DG, Libshitz HI (2001) Post-radiation sarcomas: a review of the clinical and imaging features in 63 cases. Clin Radiol 56(1): 22-29.
- Esposito E, Avino F, Di Giacomo R, Donzelli I, Marone U, et al. (2019) Angiosarcoma of the breast, the unknown—a review of the current literatura. Transl Cancer Res 8(5): 1-8.
- De Bree E, Van Coevorden F, Peterse JL, Russell NS, Rutgers EJ (2002) Bilateral angiosarcoma of the breast after conservative treatment of bilateral invasive carcinoma: genetic predisposition? Eur J SurgOncol 28(4): 392-395.
- Kadouri L, Sagi M, Goldberg Y, Lerer I, Hamburger T, et al. (2013) Genetic predisposition to radiation induced sarcoma: possible role for BRCA and p53 mutations. Breast Cancer Res Treat 140(1): 207-211.
- Jiang W, Murphy JD, Van de Rijn M, Donaldson SS (2013) Secondary breast angiosarcoma and germ line BRCA mutations: discussion of genetic susceptibility. J Radiat Oncol 2: 331-335.
- Naka N, Tomita Y, Nakanishi H, Araki N, Hongyo T, et al. (1997) Mutations of p53 tumor-suppressor gene in angiosarcoma. Int J Cancer 71(6): 952-955.
- Domfeh AB, Fichera M, Hunt JL (2006) Allelicloss of 3 different tumor suppressor gene loci in benign and malignant endothelial tumors of the head and neck. Arch Pathol Lab Med 130(8): 1184-1187.
- Heymann S, Delaloge S, Rahal A, Caron O, Frebourg T, et al. (2010) Radio-induced malignancies after breast cancer postoperative radiotherapy in patients with Li-Fraumeni syndrome. Radiat Oncol 5: 104.
- García Novoa A, Acea Nebril B, Bouzón Alejandro A, Cereijo Garea C, Antolín Novoa S (2019) Angiosarcoma radio inducido de mama en paciente con síndrome de Li-Fraumeni. Cir Esp 97(2): 114-116.
- Thibodeau BJ, Lavergne V, Dekhne N, Benitez P, Amin M, et al. (2018) Mutational landscape of radiation-associated angiosarcoma of the breast. Oncotarget 9(11): 10042-10053.
- Toesca A, Spitaleri G, De Pas T, Botteri E, Gentilini O, et al. (2012) Sarcoma of the breast: outcome and reconstructive options. Clin Breast Cancer 12(6): 438-444.
- Cao J, Wang J, He C, Fang M (2019) Angiosarcoma: a review of diagnosis and current treatment. Am J Cancer Res 9(11): 2303-2313.
- Depla AL, Scharloo Karels CH, De Jong MAA, Oldenborg S, Kolff MW, et al. (2014) Treatment and prognostic factors of radiation-associated angiosarcoma (RAAS) after primary breast cancer: a systematic review. Eur J Cancer 50(10): 1779-1788.
- Buehler D (2014) Angiosarcoma outcomes and prognostic factors. Am J Clin Oncol 37(5): 473-479.
