 Research Article
Research ArticleAbstract
Polycythaemia Vera (PV) is considered as a serious Myelo Proliferative Neoplasm (MPN). The most characteristic feature of this disease is an abnormally high red blood cell mass and/or haematocrit because of uncontrolled erythrocyte production independent of the normal regulatory processes of erythropoiesis. Recent findings has identified that 4 proteins: PDIA6, TXND5, ERP44 and PDIA1, which belong to the Protein Disulphide Isomerase (PDI) protein family demonstrated a significant increase in abundance in the red cell membrane of PV patients when compared to the healthy controls. The root cause for the PV is identified as the mutation V617F in JAK 2. In this study we have attempted to model the protein-protein interaction of the JAK2 with above identified PDI proteins using the PRISM (Protein Interactions by Structural Matching) algorithm which bases, its predictions on structural properties of any given pair of proteins and their evolutionary conserved relationships. The analysis of four PDI proteins with respect to JAK2 resulted in one meaningful interaction in which is JAK2 and TXND5 interact via CDK5, a well-known cell cycle associated protein. This finding will shed new light on the understanding of the molecular basis of PV and lead to discovery of new drugs for this disease.
Abbreviations: PDI: Protein Disulphide Isomerase; PPIs: Protein-Protein Interactions; MPN: Myeloproliferative Neoplasm; PV: Polycythemia Vera; RCM: Red Blood Cell Mass; JAK2: Janus Kinase 2; PDIA6: Protein Disulphide Isomerase A6; ERP44: Endoplasmic Reticulum Resident Protein 44; PDIA1: Protein Disulfide Isomerase A1; BID: Binding Interface Database; RMSD: Root Mean Square Deviation; ASA: Accessible Surface Area; RASA: Relative Accessible Surface Area; CTAT: Signal Transducer and Activator of Transcription; CDK: Cyclin-Dependent Kinase 5; CNS: Central Nervous System; ROS: Reactive Oxygen Species; VRK5: vaccinia-Related Kinase 3
Introduction
Most (if not all) proteins function through interactions with proteins and other molecules in order to carry out cellular functions. It has been estimated that over 80% of proteins do not function in vivo in isolation but in complexes Berggård, et al. [1]. Protein-Protein Interactions (PPIs) are involved in almost all cellular processes: regulation of cell cycle, gene expression, cell-to- cell interactions and metabolic and developmental control Braun et al. [2]. The concept of ‘‘protein interaction’’ is generally used to describe the physical contact between proteins in which they interact via their interfaces Tuncbag, et al. [3]. Therefore, studying the interface properties of proteins will help explain their role in protein-protein interactions. The biological properties of a protein molecule depend on its physical interaction with other molecules. Therefore, studies on identifying interaction sites of proteins and knowing which proteins interact with which protein and other molecules are essential to understand better their role within the cell and the basis of many cellular processes Rao, et al. [4]. Proteins that can interact with multiple partners play central role and act as hub proteins in the network of protein–protein interactions Higurashi, et al. [5]. Due to the pivotal role protein interactions play in cellular processes, they are central in controlling mechanisms leading to healthy and diseased states in organisms Kar, et al. [6,7]. Mutations in proteins can occur in active sites, allosteric binding sites and DNA binding sites. These mutations can cause changes in proteins and affect their interactions which may lead to dysfunction of some interactions and cause diseases such as cancers Kar, et al. [6,7]. Therefore elucidation of protein interaction networks could reveal the molecular basis of diseases which in turn could provide insights into developing methods of prevention, diagnosis Kann [7,8] and also lead to development of treatment methods for diseases by identification of possible drug targets Pedamallu, et al. [9].
Considering the importance of studying protein- protein
interaction networks, there has been development of several
approaches to detect these interactions. Approaches based on
genome- wide experimental methods such as the Yeast two hybrid
test, protein chips and mass spectrometric analysis has led to
the detection of numerous interactions. Recently a number of
computational approaches have been developed for the prediction
of protein-protein interactions. Computational methods for the
prediction of PPIs provide a fast and inexpensive alternative to
complement experimental efforts. Computational interaction
studies can be used to validate experimental data and to help select
potential targets for further experimental screening Shoemaker,
et al. [10]. PRISM (Protein Interactions by Structural Matching)
is a web server that can be used to explore protein interfaces
and predict protein–protein interactions Ogmen, et al. [11]. The
algorithm in PRISM principally seeks pairs of proteins that may
interact in a dataset of protein structures (target dataset) by
comparing them with a dataset of interfaces (template dataset)
which is a structurally and evolutionarily representative subset of
biological and crystal interactions present in the Protein Data Bank
(PDB) Berman, et al. [12]. PRISM consists of a web interface to the
dataset of interfaces and target structures including a summary of
the proteins the interface belongs to (with cross-references to other
biological databases where available), similarity matching results,
solvent accessible surface area calculation results on a residuelevel
scale, interface visualization of the protein using both static
images and an interactive interface viewer implemented using a
browser plug-in.
Polycythemia Vera (PV) is considered as a serious
Myeloproliferative Neoplasm (MPN). The most characteristic
feature of this disease is an abnormally high Red Blood Cell Mass
(RCM) and/or hematocrit because of uncontrolled erythrocyte
production independent of the normal regulatory processes of
erythropoiesis. This leads to hyperviscosity and an increased risk of
thrombosis Stuart, et al. [13]; Barbu, et al. 2013; Khan, et al. 2012;
Adelet, et al. 2013. The median age of patients diagnosed with PV
is 60 years, although it can occur in persons in all age groups. The
median survival in untreated symptomatic patients after diagnosis
is reported to be in the range of 6 to 18 months while it is more than
10 years for treated patients who highlight the seriousness of PV
Stuart, et al. [13]. PV occurs with a slight predominance in men and
the incidence of PV is 2-3 per 100,000 persons per year. At cellular
level the cause of polycythemia is reported to be due to increased
proliferation or decreased apoptosis of erythroid progenitors, or
due to delayed erythroid differentiation with an increased number
of progenitor cell divisions Prchal, 2001. Investigations focusing
on genetic basis of development of PV have revealed the presence
of valine to-phenylalanine mutation at amino acid 617 (V617F)
in the Janus Kinase 2 (JAK2) tyrosine kinase gene (referred to as
JAK2-V617F mutation) in a large majority of Polycythemia Vera
(PV) patients Teffreri, et al. [14], Lippertet, et al. 2006. Expression
analysis have indicated that more than 95% of PV patients had
shown expression of JAK2 gene with V617Fmutation Tefferi, et
al. [14]; Lippert, et al. 2006. Patientswith PV carry the somatic
JAK2V617F mutation in their hematopoietic cells which causes
activation of JAK2 tyrosine kinase resulting in an increase in the
phosphorylation activity of JAK2. This elevated kinase activity
of JAK2 which in turn promotes the spontaneous cellular growth
and induces uncontrolled erythrocytosis in a mouse model James,
et al. 2005; Pargadeet, et al. 2006; Baxter, et al. [15]. However, the
role of the JAK2 V617F mutation inhuman PV pathogenesis and its
impact on hematopoiesis is not fully understood and remains to be
determined Jamieson, et al. 2006.
Kottahachchi, et al. [16] employed a mass spectrometrybased
quantitative proteomics approach for the analysis of
erythrocyte membrane proteins in PV patients with the JAK2
V617F mutation. It was found that 4 proteins: Protein Disulphide
Isomerase A6 (PDIA6), Thioredoxin domain- containing Protein
5(TXND5), Endoplasmic Reticulum Resident Protein 44 (ERP44)
and Protein Disulfide Isomerase A1 (PDIA1), which belong to the
Protein Disulphide Isomerase (PDI) protein family demonstrated
a significant increase in abundance in the PV patients with the
JAK2 V617F mutation when compared to the healthy controls. It
is reported that these PDIs in PV patients functionally contributing towards alleviating oxidative stress and apoptosis in erythrocytes in
PV patients Andreu, et al. 2012. Thispossible functional role of PDIs
in protecting erythrocytes against oxidative stress and apoptosis
could lead to an increase in erythrocyte cell mass which is a feature
in PV Kottahachchi, et al. [16]. With this background, in this paper
we attempt to determine interactions between JAK2 protein PDI
proteins in erythrocyte membranes in PV patients employing the
protein interaction prediction tool, PRISM.
Methodology
In this study, we used high performance prediction PRISM algorithm to analyse the possible interactions between JAK2 and PDI proteins. For clarity purposes the methodology used in this study is shown in the work flow plan. The PRISM algorithm uses the rationale that if particular surface regions of any two proteins are spatially similar to the complementary partners of a known interface, in principle these two proteins can interact with each other via these regions. The prediction algorithm uses a template interface dataset and a target single protein structure dataset to predict such potential interactions between target proteins. The description of these two sets and the details of the PRISM algorithm are clarified in the following sections.
Protein Interactions by Structural Matching (PRISM)
In particular proteins their interfaces are evolutionary more conserved than other surface regions of the proteins. In this case The PRISM algorithm Tuncbag, et al. [3] uses two data sets to model interactions, the template data set and the target data set Baspinar, et al. [17]. The target data set contains the surface regions of the two proteins that we want to model the interaction between, i.e. JAK 2 and the PDI protein s. For this interaction model we considered only binary interactions between PDI proteins and JAK2. In other words we only considered only one PDI protein with respect to the JAK2. Therefore at a given time target dataset would be surface regions of the JAK2 and the respective. PDI protein. The analyses were made using the default settings provided with the template data set from the PRISM web server. The template data set Cukuroglu, et al. [18] consists of known, available structures from the PDB and their interfaces. The generation of the template set can be made using various physical parameters, such as atomic distances and solvent accessibilities, and modelled using Voronoi diagrams. This generated template dataset is used for structural matching with the targets provided. In this structural matching a number of parameters have been considered and a unique scoring system is used to select the optimum structural matching between the given target protein surfaces and the templates.
The Stepwise Execution of PRISM Algorithm and Theoretical Explanation
Preparation of the Template Dataset and Hotspot Generation: The method of atomic distances is used to generate the template data set. To define the interfaces of the template structures, two types of residues are defined in each chain: “interacting residues” and “nearby residues”. Two particular residues will be considered interacting residues if any two atoms from them are closer than the sum of their van der Walls radius plus 0.5 Å. A specific residue will be a nearby residue if it is a noninteracting residue and it has a C alpha atom which is positioned less than distance of 0.5Å from an interacting residue in the same chain. The accurate definitions of interacting and nearby residues are significantly important in the process of designing the interface architecture of a protein. The default template set contains 1036 structurally non redundant heterodimer interfaces. The hotspots of each template and target proteins were generated through the HOTPOINT web server Tuncbag, et al. [19]. Hotpoint is based on a few simple rules concerning solvent accessibility and the energetic contribution of residues. The thresholds of the model are adjusted according to a data set composed of 150 experimentally mutated alanine residues of which 58 residues are hot spots and 92 residues are non-hot spots. The interface residues, whose mutations change the binding free energy by at least 2.0 kcal/mol, are considered as experimental hot spots. If the mutation results in a change <0.4 kcal/mol, the residue is labelled as an experimental non-hot spot. The independent test set is derived from Binding Interface Database (BID), composed of 112 residues (of which 54 residues are hot spots and 58 residues are non-hotspots).
Surface Extraction: In this step the resultant surface area of the target proteins is extracted. Initially the method checks whether the provided target protein is a multimer or a monomer. If the target is a multimeric protein it is resolved into its respective domain chains. If the target chain has DNA or RNA structures they are not considered in this algorithm. The homologies of the target chains are initially accounted. The algorithm uses the remote access to the Naccess service for the surface area calculation. The Naccess algorithm rolls a virtual solvent probe across the target surfaces, with the radius of a solvent particle taken as 1.4 Å. Therefore the depth or height of the rolled solvent probe will be the same. The path gained by the centre of the probe gives the accessible surface area. The protein surface is the shell around the entire monomer surface. The surface regions in the target data set are extracted based on the relative accessible surface area of the residues. If the relative accessibility (i.e. the accessible surface area relative to that of the residue in an extended conformation) of a residue is more than 15%, it is labelled as a surface residue. Thereafter, the nearby residues among surface residues are extracted. They are used to provide the structural scaffolds of the protein surfaces.
Structural Alignment and Transformation Filtering: The complementary partners of the template interfaces are generated and each respective template interfaces aligned with the target surfaces using the multiport engine Shatsky, et al. 2004. This docking engine algorithm is based on the spatial similarities of amino acid coordinates while disregarding the orders of residues in the chain. The latter consideration is made because the template interfaces and target surfaces do not consists of contiguous chains. The ten best sub structural matches are generated by Multiprot Shatsky [20]. The maximal Root Mean Square Deviation (RMSD) value is set to two Å and the minimal match size is set to five residues. Multiport performs structural alignment in two steps, known as local fragment alignment and global alignment. As we have only considered pairwise alignment in this case, Multiprot first selects one molecule as stable and the other one as pivoted. This is then processed for all the possible combinations and those that are below the predefined r.m.s.d values of their 3D Euclidean transformations are considered structurally similar. The core alignment task was done using C alpha atoms. Finally, a global alignment step was performed to search for the largest structural cores between the aligned molecules. A combination of fragments is selected heuristically because finding the optimal combination is an NP-hard problem. When a unique combination is obtained, the similarity in that combination is calculated by means of (RMSD) values.
The transformation and the filtering are made by the checking of structural matching thresholds. The threshold is set to 50% in this analysis. A total of 50% of the residues in the template domain should match with the provided surfaces for the algorithm to proceed. The numbers of matching residues should also be at least 15. Transform target proteins on their similar template interfaces to form the complex structure. The transformation matrix for each matching is generated and each target protein is transformed onto the corresponding template interface partner. In this case entire target structure is considered instead of the surface region. If two partners have more than five spatially colliding residues, the match is eliminated. Side chain clashes are also neglected in the analysis. To guarantee the correct matching, the results are checked to establish whether there are at least five contacts between matching residues of the complementary partners of the template interface.
Flexible Refinements: The Hydrogen atoms are added to the transformed target proteins and the backbone flexibility is modelled by normal modes. In our modification to the original algorithm the first 50 modes of each protein are considered. The parameter file is prepared by selecting the larger protein chain as the receptor and the smaller one as the ligand. The restricted side chain option was not selected as the clashing interface residues are not assumed to be movable at both the receptor and ligands site. A total of 20% of the clashes between the side chain atoms are allowed. Then the Fibre dock engine Mashiach, et al. [21] was run to determine the side chain flexibility. This uses a rotamer library and finds the optimum combination of rotamer with the lowest total energy. CHARMM (Chemistry at Harvard Macromolecular Mechanics) force field is used for the calculation of energy. Monte Carlo iterations are applied for minimization of backbone conformation along ten normal modes.
Results
Initially, in this study all four proteins (PDIA6, TXND5, ERP44 and PDIA1) proteins were tested as potential partners of JAK2 deploying PRISM. However, PDIA1 and PDIA6 when analysed yielded a very clear unambiguous response indicating there is no interaction between PDIA1 and PDIA6 with JAK2. ERP44 protein when tested revealed three potential interactions with JAK2 through three protein domains which had the following PDB Ids: 2j23, 10BB, and 3ZRJ. These three interacting proteins when, searched using Uniprot, PDB, and SWISS prot databases, it revealed that, none of them were reported to be found in Homo sapiens. Therefore proteins that gave these interactions were not considered for further analysis. (Shown in Supplementary Data section 1). The JAK 2 and TXND5 proteins were analysed for possible interaction deploying the PRISM algorithm. This analysis revealed that TXND5 indicated one interaction with JAK2 throughCDK5 protein which acts as an intermediate protein. The PRISM algorithm which bases on structural and evolutionary similarity for its prediction on protein interactions identified considerable structural similarity between JAK2 and TXND5. Structural Alignment of JAK2 (PDB ID: 2B7A) and TXND5 (PDB ID: 3UJ1. Initially, possible interaction between JAK 2 and TXND 5 was studied based on the analysis of their sequences. However analysis of sequence between these proteins revealed that there is low sequence similarity. Low sequence similarity between JAK 2 and TXND 5 is indicative that they are highly unlikely to interact directly Pearson [22]. In order to discover any possible interactions between JAK 2 and TXND5 analysis of structural alignment was carried out employing multiprot algorithm which bases the analysis on 3D structural properties of JAK 2 and TXND5 proteins. The output of the structural alignments of two proteins is based on the RMSD (root mean squared values) which is a measure of their divergence from one another. The RSMD value was 1.98 in this alignment. The structural alignment gives superimposition of the atomic coordinates. The minimum information generated by each respective structural alignment is a set of superimposed coordinates for each input structure. The Table 1 shows the structurally aligned residues of JAK2 with respect to its matching residues in TXND5.When aligning two structures the side chain’s atoms were not taken into account. Analysis of results of multiprot algorithm as shown in Table 2 revealed a high structural alignment between sequentially unrelated (JAK2 and TXND5 proteins. This revealed these two proteins have a possibility of interacting with each other via a complex irrespective of their low sequence similarity. In this protein complex the target protein (TXND5) is predicted to act as a putative binding ligand to the protein complex.
Hot Spot Generation and Interface Defining of JAK2 (PDB ID: 2B7A)
The hotspots were generated because of the interactions among two interfaces occur through them. The input for the HOTPOINT web server was the pdb format file of JAK2 which plays the key role in patho physiology of PV. Along with the pdb formatted file, the chain identifiers of the domains of JAK2were also employed as an input parameter for the analysis in order to define the interfaces. The output of the HOTPOINT server is given as a table (Table 1) which consists of the interface residues and their features. The interface residues were tabulated with chain names, one- letter residue names, and residue numbers, their relative ASA (Accessible Solvent Area), relative ASA in monomer and total pair potentials. In the last column of the table (Table 1), the prediction is presented as H (hot spot) or NH (non-hotspot). The identified hot spots are highlighted (Table 1).
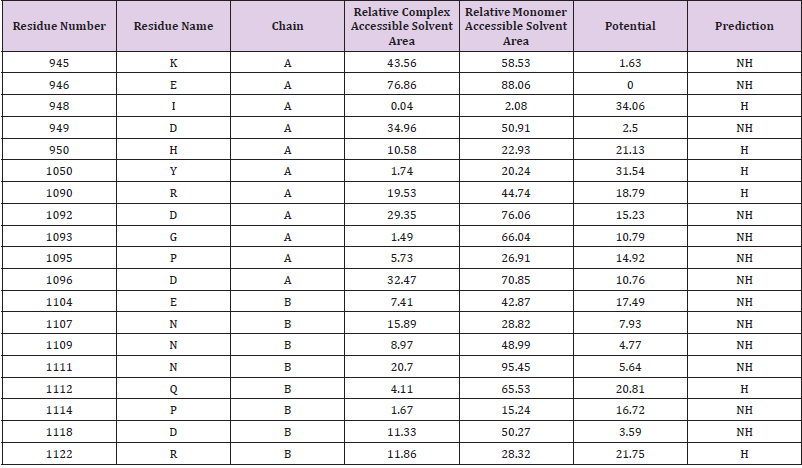
Table 1: The Hotspot residues of the JAK2 (PDB ID: 2B7A) and their accessible surface areas.
The identified hot spots are in Jmol visualization in the interface between the chain A and chain B of the JAK2 protein is illustrated. The hotspot residues present in chain A and chain Bare separately distinguished from their interface residues. The cartoon illustrations of the potential ligands that can be bound to each interface of the JAK2 are illustrated respectively in light blue and yellow.
Construction of the Interaction Complex by Searching Through Template Data Set
The construction of the interaction complex with two target proteins JAK2 (PDB ID: 2B7A) and TXND5 (PDB ID: 3UJ1) that would facilitate the interaction was initiated after the identification of interface with hot regions of the JAK2 and considering TXND5 as a putative binding ligand to the complex. Respective two targets were matched against the template data set to construct the interaction complex. The interfaces and hotspots of the template proteins were extracted by the database itself and their hotspots were defined by PRISM in the similar way as was done for the target proteins. The construction of an interaction complex using template dataset according to the PRISM protocol revealed that the two target proteins JAK2 (PDB ID: 2B7A) and TXND5 (PDB ID: 3UJ1) could build up a complex through the protein CDK5 (PDB ID: 1UNH). CDK5 a well-known cell cycle associated protein that has protein cyclin kinase activity. The output from the hotspot analysis of CDK5 is illustrated in Table 3. Here the interface residues were tabulated with chain names, one-letter residue names, and residue numbers, their relative ASA in complex, relative ASA in monomer and total pair potentials. In the last column of the table (Table 2), the prediction is presented as H (hot spot) or NH (non-hotspot). Background of the predicted hot spots in the table is highlighted with cyan, dark red, orange and red the Pymol visualization four types of hot spots in CDK5 which is shown in the Table 3, which are distinguished by their respective colours. The illustrations show two domains of the CDK5 and its extracted hot regions from the surface. The colour of the each hot spot denotes its type. There are four distinguishable hot regions in the both domains of the CDK5. The two domains together compose the motif that facilitates the binding of the external ligands, in this case the two target protein complexes JAK2 (PDB ID: 2B7A) and TXND5 (PDB ID: 3UJ1). The residues that contribute to build up the same type hot spots are distributed between two domains. As an example the hot spots that denoted by the light blue colour is from both domain B and E (residue 76 is from domain B and residue 243 is from domain E). The binding motifs in CDK5 for the two target proteins JAK2 (PDB ID: 2B7A) and TXND5 (PDB ID: 3UJ1) are located in its B and E domains.
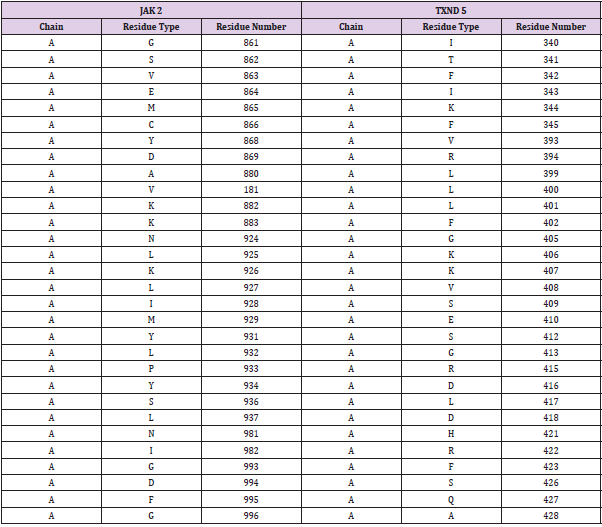
Table 2: The Structural alignment results of the two target proteins with their residue numbers and name. The table shows the structurally fitting residues of JAK2 and TXND5 they are shown side by side. As an example the residue number 861 of the JAK2 is structurally aligning with the residue number 340 of the TXND5.
The calculated fire dock energy data clearly showed how CDK 5 is the most suitable intermediate for this complex between JAK2 and TXND5 being an outlier among other tested proteins. The Selection of the most suitable protein that would fit in as the intermediate protein to form the complex was based on fire dock energy score Andrusier, et al. [23]. The fire dock energy score which is based on the aggregated approximation of the binding free energy function. Aggregated free energy also known as interface energy is sum of several energetic factors. In this analysis the benchmark value for the fire dock energy score was set as 1.0 Chen, et al. [24] and ranking of fire dock energy scores demonstrated that among the potential candidate proteins analysed, only CDK5 with a fire dock score of 1.81 exceeded the benchmark and was the unambiguous choice as intermediate protein. (Other disqualified proteins and the calculation of fire dock energy scores are given in the Supplementary Data Section 4 for further information.) In addition to the residue type and its location, the PRISM algorithm reveals the Accessible Surface Area (ASA) and the Relative Accessible Surface Area (RASA) in both the monomer and the complex states. These parameters play a significant role in deciding their pair potentials and binding energies. The ASA is the surface area that is exposed to the outside of a certain residue and RASA is the ratio between ASA and the total surface area of the residue. The final three member protein interaction complex is illustrated, showing how the two target proteins JAK 2 (PDB ID: 2B7A) and TXND5 (PDB ID: 3UJ1) interact through CDK5 (PDB ID: 1UNH). The two targets fit into the motif between domain E and B of CDK5 giving rise to the overall protein complex.
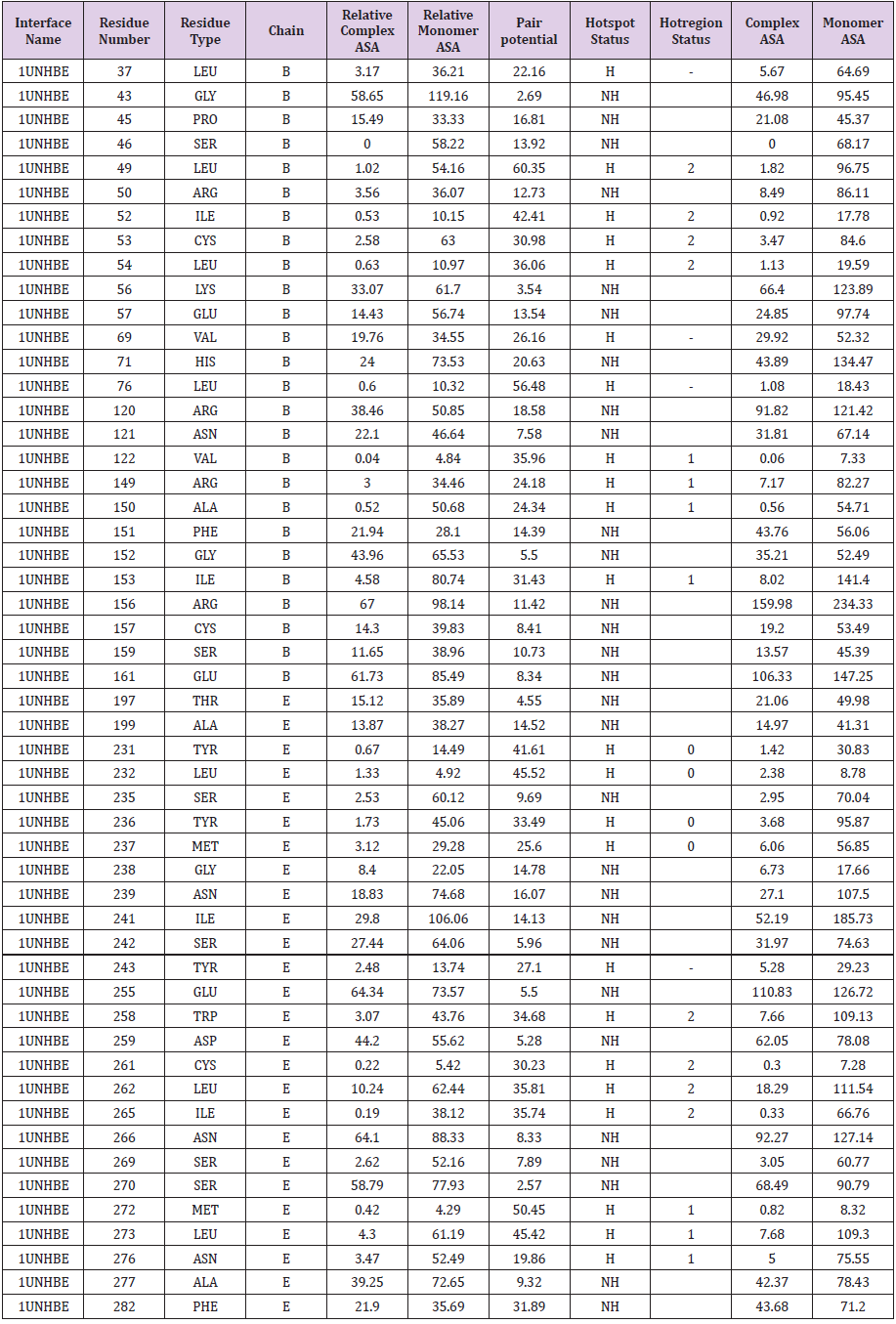
Table 3: The structural properties of hot spots residues that (the respective hot spots are categorized in to five different groups coloured by cyan, dark red, orange and red) which are contributing to hot regions in the CDK 5 (PDB ID: 1UNH).
Discussion
PPI occur as a result of physical contact between proteins
and their interacting partners. These interactions occur via their
interfaces. Therefore, studying the interface properties of interacting
protein molecules provide useful insights to the molecular
mechanisms involved in cellular functions and biological processes.
Furthermore, interactions between proteins carrying mutations
that result in dysfunctional protein interactions is considered to be
the common mechanism that lead to diseases, including cancer Kar,
et al. [6,7]. Hence studying the underlying protein interactions is
likely to enhance our understanding of the molecular mechanism
that bring about diseases. In the present study, the focus was to
predict protein-protein interactions, centred on JAK2 protein with
V617F mutation. This mutation is known to result in the production
of a constitutively activated JAK2 protein, which is considered to
play an important role in the development of Polycythaemia Vera
Tefferi, [14]; Gnanasambandan, et al. 2010. A computational
approach was adopted to predict PPIbetween JAK2 protein and four
proteins [PDIA1 PDIA6, TXND5, and ERP44] belonging to Protein
Disulphide Isomerase (PDI) protein family. The choice of these PDI
proteins for analysis possible protein interaction with JAK2 protein
was based on the findings of Kottahachchi, et al. [16] where, a high
abundance of these PDI proteins were identified and quantified
by a LC–MS/MS based quantitative proteomics approach during
analysis of erythrocyte membrane proteins from PV patients with
the JAK2 V617F mutation. Prediction of interactions of JAK2 protein
with any member of above stated PDI protein family would enable
us to gain a better understanding of the mechanism underlying this
disease at the molecular level.
The rationale of PRISM is as follows: if complementary partners
of a template interface are similar to surface regions of any two
proteins, these two proteins can interact with each other through
these regions. When searching for similar spatial motifs on the target
protein surface, PRISM considers both geometric complementarity
detected by structural alignments and evolutionary conservation
of hot spots. No sequence similarity is used Tuncbagetal, 2011.
The PPIof fourPDI proteins erythrocytePDIA1 PDIA6, TXND 5 and
ERP44) with JAK2) were analysed using the PRISM. No interactions
were predicted for PDIA1 and PDIA6 () with JAK2. ERP44) gave
three interactions with JAK2) through three protein domains with
the following PDB Ids; 2j23 AB, 10BB AB and 3ZRJ AB. However,
there is no evidence of the presence of these three interacting
proteins in Homo sapiens; thus they were not considered for
further analysis. One interaction was predicted for TXND5) with
JAK2 and was subjected to further analysis. Structural alignment
of two target proteins JAK2 and TXND5revealed that there is
considerable structural similarity of 62% (RCSB PDB) between
the target proteins despite their low sequence similarity of 11.9%.
Therefore it can be deduced by structural alignment of two target
proteins JAK2 and TXND5 that they are having a close evolutionary
relationship and hence they are more likely to interact with each
other through a complex.JAK2, which is a multimeric protein, was
subjected to analysis initially in our study in order to discover
all the possible matches from the template data set as there is a
high probability to find a matching partner from the template
data set for a multimer rather than a monomer which has lesser
number of potential matches. Using Hot point web server the hot
spots of JAK2 were generated based on few simple rules such as,
if a particular interface residue results in a binding energy more
than 2.0kcal/mol it was categorized as a hot spot and if the binding
energy of a particular surface residue was less than 2.0kcal/mol it
was categorized as a non-hotspot by default Lucet, et al. [25]. At
the same time the rules that define surface residues also applies
when generating hotspots. In addition, interface residues were
identified from the given structure using a threshold level of relative
accessible solvent area (RASA) for a given residue (Baspinar et al.,
2014). If the RASA for a given residue is more than or equals to 20% it was categorized as an interface residue. This analysis led to the
identification of six distinct hotspot residues in JAK2 protein that
can be considered to have the highest probability of interacting
with another protein.
The computational approach which resulted in the generation
and determination of accessibility of the identified hotspots for
possible interaction in extracted surfaces of the two target proteins
JAK2 and TXND5 revealed that they could not interact directly.
However, subsequent search of the template data set predicted
that JAK2 and TXND5 could interact with each other facilitated
by an intermediate protein identified as cell cycle related protein
CDK5. The intermediate proteins were selected according to the
ranking of fire dock energy score Andrusier, et al. [23]. An energy
score benchmark was defined and the only protein qualified to
exceed the benchmark was CDK5 As was discussed analysis of
structural aspects of proteins JAK2, CDK5 and TXND5 identified
the above mentioned three member interacting protein complex.
Analysis of domains of proteins not only allows one to determine
the nature of their interactions but also provides clues to determine
the function of the proteins as well Vogel, et al. 2004. In order to
elucidate possible functional relationships among these three
members of the protein complex, functional role of domains in
JAK2, CDK5and TXND5 were analysed based on information
available in the literature. The properties of a proteins are largely
determined by its three dimensional structure. Therefore it is very
likely to have close functional interactions among above discovered
three member protein complex as they demonstrate d considerable
strong structural interactions between each other Hvidsten, et al.
2009.
Protein kinases with conserved catalytic kinase domains make up one of the largest ‘super families’ of eukaryotic proteins. Protein kinases play a crucial role in the integration of signal transduction networks, and the signalling pathways of mammalian cells Zhang, et al. [26]. Among different protein kinases, Janus kinases (JAKs) are a family of non-receptor protein tyrosine kinases involved in signalling cascades initiated by various cytokines, interferon, and growth factors. It is found that kinase activity of Janus kinases (JAKs) are essential for activation of signalling mediated by cytokine receptors that lack catalytic activity Myers, et al. [27]. Upon hormone binding, JAKs phosphorylate tyrosine residues in the receptor cytoplasmic domains and in JAKs themselves leading to recruitment and activation of downstream signalling proteins such as Signal Transducer and Activator of Transcription (STAT) pathway Klingmuller, et al. [27,28]. The JAK2/STAT signalling pathway affects cellular activities, such as cell proliferation, migration, growth, differentiation and cell death Duan, et al. [29]. Analysis of functional role of JAK2 protein with V617F mutation is found to undergo constitutive activation of JAK2 as a result of auto phosphorylation without the binding of hormone erythropoietin. This results in the loss of cell proliferation regulatory function of JAK2/STAT pathway and induces inappropriate cytokineindependent proliferation of hematopoietic cells. Which in turn results in uncontrolled erythropoiesis in PV that lead to the accumulation of morphologically normal red cells Alabdulaali, et al. 2009; Baxter, et al. [15].
Cyclin-dependent kinase 5 (CDK5) which act as the intermediate protein the of the three member protein complex that is being the focus of this study. CDK5is a highly conserved member of the small serine/ threonine cyclin-dependent kinase (CDK) family. Members of this family are found to be important in regulation of cell growth, differentiation and cell death Zho, et al. 2002 and are expressed predominantly in post mitotic cell populations Kanungo, et al. [30]. Recent findings suggest that CDK5 is an unusual member of the CDK family as it is reported to promote cell survival by activating anti-apoptotic proteins Lalioti, et al. [31]. The most studied function of CDK5 is found to be its role in regulation of the cytoskeleton architecture of the Central Nervous System (CNS). Furthermore, it is reported that CDK5, to be predominantly activated in the neurons because the expression of its activators, p35 and p39, are restricted to neuronal tissue Song, et al. 2016. Evidence suggests that CDK5 is an unusual member of the CDK family which is found to promote cell survival by activating anti-apoptotic proteins involved in promoting neuronal survival during oxidative stress. Recent findings of Song et al 2016 show that oxidative stress is caused by an imbalance between Reactive Oxygen Species (ROS), such as hydrogen peroxide (H2O2) production and antioxidant defence mechanisms. This imbalance results in an increase in extracellular signal-regulated kinase 1/2 (ERK 1/2) activation and unusually sustained ERK activation is found tolead to neuronal cell death. Song, et al. (2016) show that oxidative stress-induced cyclin-dependent kinase 5 (CDK5) activation stimulates neuron protective function in neurons via phosphorylation of vaccinia-related kinase 3 (VRK3) at Ser 108 which in turn suppresses H2O2 induced prolonged ERK activation and promote survival of neuronal cells. These findings demonstrate that CDK5 play an important anti apoptotic function in prolonging the lifespan of neuronal cells exposed to oxidative stress. However a similar anti apoptotic function of CDK5 in other cells is not reported in the literature other than neuronal cells. Both CDK5 and JAK2 share common protein kinase domains and kinase like domains. CDK5 mainly consists of Serine/threonine protein kinase domains and includes with two main chains, A and B Maccioni, et al. [32].
TXND5 is the third protein of the three member protein complex which has been the focus of the present study [33-35]. TXND5 is a member of the PDI protein family in which members have been found to contain at least one thioredoxin (Trx) domain. Erythrocytes are constantly exposed to oxidative stress as they are rich in oxygen and have an extensive array of antioxidants to counter this level of stress Hattangadi and Lodish, 2007. Excess oxidative stress which causes elevated levels of ROS (reactive oxygen species) such as superoxide O–2, hydroxyl radical OH, and H2O2 are constantly generated in cells during intracellular metabolism and in response to environmental stimuli [36-39]. ROS is known to damage macromolecules vital for cellular functions and which could result in apoptosis, cell cycle disruption and necrosis Halliwell, 2007; Trachootham, et al. 2008. Oxidative stress is known to induce thioredoxin expression that provides protection against the stress and which also reported to have the ability to delay apoptosis Powis and Kirkpatrick, 2007; Qu, et al. 2011; Valko, et al. 2006; Karlenius, et al. 2010; Flores, et al. 2012. In PV patients erythrocytes are exposed to very high levels of oxidative-stress conditions and increased abundance of thioredoxin domain containing PDI proteins such as TXND5 have been reported Kottahachchi, et al. [16]. It is suggested that PDI proteins including TXND5 could act as an inhibitory factor for oxidative stress and enhance the tolerance of erythrocytes to oxidative stress and delaying oxidative stress induced apoptosis Kottahachchi, et al. [16]. This process may provide a mechanism for prolonged life span of erythrocytes and could contribute towards in high erythrocyte mass found in PV patients [40-43].
The above evidence reveal that CDK 5 is major component in neuronal cell apoptotic process and lays a foundation to build up the implication that CDK5 structurally as well as functionally interacts with TXND5 which is also having the major function as negative regulation of the apoptotic process [44-47]. Furthermore all three member complex of the complex JAK2, CDK5 and TXND5 possess negative regulation of the apoptotic process as their functional annotations. JAK2 and CDK5 both having evidences for negative regulation of the neuronal apoptosis and TXND5 is referred as generally negative regulation of the apoptosis Bruneel, et al. 2005. Proteomics of human umbilical vein endothelial cells applied to etoposide-induced apoptosis. It is believed to be that there is a very high potential of these JAK2 and CDK5 is involved with the negative regulation of the apoptosis in the red blood cells also [48-50]. Interactions between proteins play an essential role in the proper functioning of living cells and also PPIs mediate essentially all biological processes. We have elucidated a pathway that two target proteins JAK2 and TXND5 interact through CDK5. To form interactions between two proteins, they should share common structural similarities as well as common functional properties [51-53]. The three proteins have structural similarities, such as the presence of thioredoxin like domains (Trxs) and these Trxs act against oxidative stress induced apoptosis of the cells. Patients with PV carry the somatic JAK2V617F mutation in their hematopoietic cells [54-57]. This mutation results in constitutive activation of JAK2 tyrosine kinase which leads to an increase in the phosphorylation activity of JAK2, which in turn promotes the spontaneous cellular growth and induces uncontrolled erythrocytosis [58-61]. JAK2 and CDK5 being kinases that regulates the cell cycle and TXND5 carrying anti apoptotic function the JAK2-CDK5-TXND5 pathway could additionally supports this hypothesis that over production of red blood cells in PV patients and also their prolonged life span [62-65].
In this study PRISM, a template based, computational protein– protein interaction prediction tool was successfully employed in which it predicted JAK2 protein interacting with TXND5 via CDK5. PRISM for its predictions as stated earlier, uses interfaces of large number of non- redundant protein templates extracted from PDB. The analysis deploying a template data set comprising of 22604 protein interfaces with known interactions for its predictions on protein – protein interactions, invariably enhances both the reliability and the accuracy of the results obtained pertaining to JAK2 protein interacting with TXND5 via CDK5 [66-68]. In an attempt to validate the overall accuracy of PRISM, it was compared with STRING database. The F measure calculated, demonstrated that predictions on PPI computationally generated relying on structural properties of proteinsby PRISM and PPI based on experimental data in STRING are comparable, and both methods have similar performance as tools in elucidating protein-protein interactions [69]. Furthermore, PRISM appears to be an appropriate computational tool which has been reported as a web server which enables efficient, protein –protein interactions with high accuracy Basipinar, et al. 2014.
Conclusion
On the basis of in-silico analysis of putative interaction sites of a panel of differentially abundant proteins identified in PV erythrocytes we were able to propose a protein complex that included the TXND5-CDK5-JAK2. On the basis of the similar properties of the complex components their activity in PV erythrocytes could involve the resistance of oxidative stress induced apoptosis. Identifying a pathway related to oxidant-induced apoptosis may therefore uncover novel therapeutic targets for PV. However, prior to implement this, there should be an experimental validation of the results that achieved by computational methods.
References
- Berggård T, S Linse, James P (2007) Methods for the detection and analysis of protein-protein interactions. Proteomics 7(16): 2833-2842.
- Braun P, Gingras AC (2012) History of protein-protein interactions: from egg-white to complex networks. Proteomics 12(10): 1478-1498.
- Tuncbag N, Gursoy A, Nussinov R, Keskin O (2011) Predicting protein-protein interactions on a proteome scale by matching evolutionary and structural similarities at interfaces using PRISM. Nat Protoc 6(9): 1341-1354.
- Rao VS, Srinivas K, Sujini GN, Kumar GNS (2014) Protein-Protein Interaction Detection: Methods and Analysis. International Journal of Proteomics 2014: 147648.
- Higurashi M, Ishida T, Kinoshita K (2008) Identification of transient hub proteins and the possible structural basis for their multiple interactions. Protein Science 17(1): 72-78.
- Kar G, Gursoy A, Keskin O (2009) Human Cancer Protein-Protein Interaction Network: A Structural Perspective. PLoS Comput Biol 5(12): e1000601.
- Gonzalez MW, Kann MG (2012) Chapter 4: Protein Interactions and Disease, PLOS Computational Biology 8(12): e1002819.
- Kann MG (2007) Protein interactions and disease: computational approaches to uncover the etiology of diseases. Briefings in Bioinformatics 8(5): 333-346.
- Pedamallu CS, Posfai J (2010) Open source tool for prediction of genome wide protein-protein interaction network based on ortholog information. Source Code for Biology and Medicine 5(1): 8.
- Shoemaker BA, Panchenko AR, Bryant SH (2006) Finding biologically relevant protein domain interactions: conserved binding mode analysis. Protein Sci 15(2): 352-361.
- Ogmen U, Keskin O, Aytuna AS, Nussinov R, Gursoy A (2005) PRISM: protein interactions by structural matching. Nucleic Acids Research 33: W331-W336.
- Berman HM, Westbrook JZ, Feng Z, Gilliland G, Bhat TN, et al. (2000) The Protein DataBank. Nucleic Acids Res 28(1): 235-242.
- Stuart BJ, Viera AJ (2004) PolycythemiaVera. American Family Physician 69(9): 2139-2144.
- Tefferi A (2006) Classification, diagnosis and management of myeloproliferative disorders in the JAK2V617F Era. American Society of Hematology 2006(1): 240-245.
- Baxter EJ, Scott LM, Campbell PJ, Clare East, Nasios Fourouclas, et al. (2005) Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 365(9464): 1054-1061.
- Kottahachchi D, Gooneratne L, Jayasekera A, Muth-Pawlak D, Moulder R, et al. (2015) Quantitative analysis of the erythrocyte membrane proteins in polycythemiavera patients treated with hydroxycarbamide. Eupa Open Proteomics 7: 43-53.
- Baspinar A, Cukuroglu E, Nussinov R, Keskin O, Gursoy A (2014) PRISM: a web server and repository for prediction of protein-protein interactions and modelling their 3D complexes. Nucleic Acids Res 42(W1): W285-289.
- Cukuroglu E, Attila Gursoy, Ruth Nussinov, Ozlem Keskin (2014) Non-Redundant Unique Interface Structures as Templates for Modeling Protein Interactions. PLOS one 9(1): e86738.
- Tuncbag N, Keskin O, Gursoy A (2010) HotPoint: Hot spot prediction server for protein interfaces. Nucleic Acids Research 38(2): 402-406.
- (2002) Shatsky MultiProt-A Multiple Protein Structural Alignment Algorithm. Lecture Notes in Computer Science 2452: 235-250, Springer Verlag, 2002. Workshop on Algorithms in Bioinformatics, WABI 2452: 235-250.
- Mashiach E, Nussinov R, Wolfson HJ (2010) Fiber Dock: Flexible induced-fit backbone refinement in molecular docking. Proteins: Structure, Function and Bioinformatics 78(6): 1503-1519.
- Pearson WR (2014) An Introduction to Sequence Similarity (“Homology”) Searching. Curr Protoc Bioinformatics, p. 1-9.
- Andrusier N, Nussinov R, Wolfson H J (2007) FireDock: Fast interaction refinement in molecular docking. Proteins 69(1): 139-159.
- Chen R, Mintseris J, Joël Janin, Weng Z (2003) A Protein - Protein Docking Benchmark. Proteins 52(1) 88-91.
- Lucet IS, Fantino E, Styles M, Bamert R, Patel O, et al. (2006) The structural basis of Janus kinase 2 inhibition by a potent and specific pan - Janus kinase inhibitor. Structure 107(1): 176-183.
- Zhang J, Luan CH, Chou KC, Johnson GVW (2002) Identification of the N-terminal functional domains of Cdk5 by molecular truncation and computer modeling. Proteins: Structure, Function and Genetics 48(3): 447-453.
- Myers MP, Andersen JN, Cheng A, Tremblay ML, Horvath CM, et al. (2001) TYK2 and JAK2 are substrates of protein-tyrosine phosphatase 1B. Journal of Biological Chemistry 276(51): 47771-47774.
- Klingmuller U, Lorenz U, Cantley LC, Neel BG, Lodish HF (1995) Specific recruitment of SH-PTP1 to the erythropoietin receptor causes inactivation of JAK2 and termination of proliferative signals. Cell 80(5): 729-738.
- Duan W, Yang Y, Yan J, Yu S, Liu J, et al. (2012) The effects of curcumin post treatment against myocardial ischemia and reperfusion by activation of theJAK2/STAT3 signaling pathway. Basic Res Cardiol 107(3): 263.
- Kanungo J, Li B, Zheng Y, Pant HC (2006) Cyclin-dependent kinase 5 influences Rohon-Beard neuron survival in zebrafish. Journal of Neurochemistry 99(1): 251-259.
- Lalioti V, Pulido D, Sandoval IV (2010) Cdk5, the multifunctional surveyor. Cell Cycle 9(2): 284-311.
- Maccioni RB, Otth C, Concha II, Munoz JP (2001) The protein kinase cdk5: Structural aspects, roles in neurogenesis and involvement in Alzheimer’s pathology. European Journal of Biochemistry 268(6): 1518-1527.
- Alanen HI, Salo KE, Pekkala M, Siekkinen HM, Pirneskoski A, et al. (2003) Defining the domain boundaries of the human protein disulfideisomerases. Antioxidants & Redox Signaling 5(4): 367-374.
- Al Lazikani B, Sheinerman FB, Honig B (2001) Combining multiple structure and sequencealignments to improve sequence detectionand alignment: Application to the SH2domains of Janus kinases. PNAS 98(26): 14796-14801.
- Bratosin D, Estaquier J, Petit F, Arnoult D, Quatannens B, et al. (2001) Programmed cell death in mature erythrocytes: a model for investigating death effector pathways operating in the absence of mitochondria. Cell Death Differ 8: 1143-1156.
- Chang X, Xu B, Wang L, Wang Y, Yan S, et al. (2013) Investigating a pathogenic role for TXNDC5 in tumors. Int J Oncol 43(6): 1871-1884.
- Dacie JV, Lewis SM (2001) Practical Haematology (9th Edn.)., Churchill Livingstone, London, UK.
- Denisov AY, Maattanen P, Dabrowski C, Kozlov G, Thomas DY, et al. (2009) Solution structure of the bb’ domains of human protein disulfide isomerase. FEBS Journal 276(5): 1440-1449.
- Dimauro T, David G (2010) Ras-induced Senescence and its Physiological Relevance in Cancer. Curr Cancer Drug Targets 10(8): 869-876.
- Droit A, Poirier GG, Hunter JM (2005) Experimental and bioinformatic approaches for interrogating protein-protein interactions to determine protein function. J Mol Endocrinol 34(2): 263-280.
- Duivenvoorden WC, Paschos A, Hopmans SN, Austin RC, Pinthus JH (2014) Endoplasmic reticulum protein ERp46 in renal cell carcinoma. PLoS One 9(3): e90389.
- Eggers JP, Grandgenett PM, Collisson EC, Lewallen ME, Tremayne J, et al. (2011) Cyclin- dependent kinase 5 is amplified and overexpressed in pancreaticcancer and activated by mutant K- Ras. Clin Cancer Res 17(19): 6140-6150.
- Ehrlich SM, Liebl J, Ardelt MA, Lehr T, De Toni EN, et, al. (2015) Targetingcyclin dependent kinase 5 in hepatocellular carcinoma-A novel therapeuticapproach. J Hepatol 63(1): 102-113.
- Feldmann G, Mishra A, Hong SM, Bisht S, Strock CJ, et al. (2010) Inhibiting the cyclin- dependent kinase CDK5 blocks pancreatic cancer formation and progression through the suppression of Ras-Ralsignaling. Cancer Res 70(11): 4460-4469.
- Filippakopoulos P, Muller S, Knapp S (2009) SH2 domains: modulators of nonreceptor tyrosine kinase activity. Current Opinion in Structural Biology 19(6): 643-649.
- Henchcliffe C, Burke RE (1997) Increased expression of cyclin-dependent kinase 5 in induced apoptotic neuron death in rat substantianigra. Neurosci Lett 230(1): 41-44.
- Horna Terrón E, Pradilla Dieste A, Sánchez de Diego C, Osada J (2014) TXNDC5, a Newly Discovered Disulfide Isomerase with a Key Role in Cell Physiology and Pathology. Int J Mol Sci 15(12): 23501-23518.
- Kalesnikoff J, Sly LM, Hughes MR, Büchse T, Rauh MJ, et al. (2003) The role of SHIP in cytokine-induced signaling.Reviews of Physiology. Biochemistry and Pharmacology 149: 87-103.
- Lang KS, Lang PA, Bauer C, Duranton C, Wieder T, et al. (2005) Mechanisms of suicidal erythrocyte death. Cell PhysiolBiochem15(5): 195-202.
- Lenherr ED, Kopp J, Sinning I (2011) Complex of ClpV N-domain with VipB peptide J Biol Chem.
- Limacher A, Glaser AG, Meier C, Schmid-Grendelmeier P, Zeller S, et al. (2007) Cross- Reactivity and 1.4-Å Crystal Structure of Malasseziasympodialis Thioredoxin (Mala s 13), a Member of a New Pan-Allergen Family. J Immunol 178: 389-396.
- Liu X, Li L, Chen ZJ, Lu Z, Shi Y, et al. (2010) Genetic variants of cyclin-dependentkinase 5 regulatory subunit associated protein 1-like 1 and transcription factor7-like 2 are not associated with polycystic ovary syndrome in Chinese women. Gynecol Endocrinol 26(2): 129-134.
- Lodge JA, Maier T, Lieb W, Hoffmann V, Strater N (2003) Crystal Structure of ThermotogaMaritima Alpha-GlucosidaseAgla Defines a New Clan of Nad+-Dependent Glycosidases. J Biol Chem 278(21): 19151-19158.
- Mapelli M, Massimilinao L, Crovace C, Seeliger MA, Tsai LH, et al. (2005) Mechanism of Cdk5/P25 Binding by Cdk Inhibitors. J Med Chem 48(3): 671-679.
- Neubauer H, Cumano A, Muller M, Wu H, Huffstadt U, et al. (1998) Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell 93(3): 397-409.
- Noh JY, Oh SH, Lee JH, Kwon YS, Ryu DJ, et al. (2010) Can blood components withage-related changes influence the ageing of endothelial cells? Exp Dermatol 19: 339-346.
- Patrick GN, Zukerberg L, Nikolic M, Delamonts S, Dikkes P, et al. (1999) Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402(6762): 615-622.
- Sam L, Liu Y, Li J, Friedman C, Lussier YA (2007) Discovery of protein interaction networks shared by diseases. Pacific Symposium on Biocomputing, pp. 76-87.
- Sandberg EM, Sayeski PP (2004) Jak2 Tyrosine Kinase Mediates Oxidative Stress-induced Apoptosis in Vascular Smooth Muscle Cells. The journal of biological chemistry 279(33): 34547-34552.
- Sarmady MW, Dampier W, Tozeren A (2011) HIV protein sequence hotspots for crosstalk with host hub proteins. PLoS ONE 6(8): e23293.
- Sun KH, De Pablo Y, Vincent F, Shah K (2008) Deregulated Cdk5 promotes oxidative stress and mitochondrial dysfunction. J Neurochem 107(1): 265-278.
- Wang Y, Ma Y, Lu B, Xu E, Huang Q, et al. (2007) Differential expression of mimecan and thioredoxin domain-containing protein 5 in colorectal adenoma and cancer: a proteomic study. Exp Biol Med (Maywood) 232(9): 1152-1159.
- Vincent EE, Elder DJ, Phillips L, Heesom KJ, Pawade J, et al. (2011) Overexpression of the TXNDC5 protein in non-small cell lung carcinoma. Anticancer Res 31(5): 1577-1582.
- Von Mering CR, Krause R, Snel B, Cornell M, Oliver SG, et al. (2002) Comparative assessment of large-scale data sets of protein- protein interactions, Nature 417(6887): 399-403.
- Yeh TC, Dondi E, Uze G, Pellegrini S (2000) A dual role for the kinase-like domain of the tyrosine kinase Tyk2 in interferon- signaling. Proc Natl Acad Sci 97(16): 8991-8996.
- Zhang A (2009) Protein Interaction Networks-Computational Analysis, Cambridge University Press, NewYork, NY, USA.
- Zhang L, Hou Y, Li N, Wu K, Zhai J (2010) The influence of TXNDC5 gene on gastric cancer cell. J Cancer Res Clin Oncol 136(10): 1497-1505.
- Zhang Q, Ahuja HS, Zakeri Z, Wolgemuth DJ (1997) Cyclin-dependent kinase 5 is associated with apoptotic cell death during developmental and tissue remodeling. Dev Biol 183: 222-233.
- Zhu Y, Lin L, Kim S, Quaglino D, Lockshin RA, et al. (2002) Cyclin dependent kinase 5 and its interacting proteins in cell death induced in vivo by cyclophosphamide in developing mouse embryos. Cell Death and Differentiation 9(4): 421-430.
