 Research Article
Research ArticleAbstract
Recent progress in RNA biology has broadened the scope of therapeutic targets of RNA drugs for cancer therapy. However, RNA drugs, typically small interfering RNAs (siRNAs), are rapidly degraded by RNases and filtrated in the kidney, thereby requiring a delivery vehicle for efficient transport to the target cells. To date, various delivery formulations have been developed from cationic lipids, polymers, and/or inorganic nanoparticles for systemic delivery of siRNA to solid tumors. This research article describes the current status of clinical trials related to siRNA-based cancer therapy, as well as the remaining issues that need to be overcome to establish a successful therapy. It, then introduces various promising design strategies of delivery vehicles for stable and targeted siRNA delivery, including the prospects for future design. The current major strategies to design delivery vehicles for systemic siRNA delivery involve the construction of multimolecular assemblies from more than dozens of monomer components, including siRNA. The success of RNAi-based cancer therapy is closely associated with tumor biology as well as architecture of delivery vehicles. Tumor cell plasticity evokes a resistance mechanism against clinical treatments, and cancer stem cells are gradually being identified as the root of cancer recurrence. New target RNA genes should be discovered to increase apoptosis in cancer cells and simultaneously reduce side effects in normal and healthy cells. Multidisciplinary research studies will guide the development of highly effective and safer RNAi-based drugs in clinical trials.
Keywords:RNAi; siRNA Delivery; Polyion Complex Micelle; Systemic Administration; Cancer Therapy
Abbreviations: GBM: Glomerular Basement Membrane; EPR: Enhanced Permeability and Retention; TEM: Transmission Electron Microscopy; FDA: Food and Drug Administration; Fc: Crystallizable Fragment; Fab: Antigen-Binding Fragment; PLD: Pegylated Liposomal Doxorubicin, PFS: Median Progression-Free Survival; PICs: Polyion Complexes; PEI: Polyethyleneimine; HA: Hyaluronic Acid; Glu: Glutamic Acid; Cys: Cysteine; Gly: Glycine; FPBA: 3-Fluoro-4-Carboxyphenylboronic Acid; LbL: Layer-By-Layer; CaP: Calcium Phosphate; BBB: Blood-Brain Barrier; BTB: Blood-Tumor Barrier
Introduction
Extracellular and Intracellular Barriers in Systemic siRNA Delivery to Solid Tumors
Various delivery vehicles have been developed for systemic delivery of therapeutic siRNA into solid tumors [1]. They need to protect vulnerable siRNA from enzymatic degradation and avoid rapid renal filtration as well as entrapment by phagocytes, and further extravasate from blood to tumor tissues (Figure 1A). Once siRNAs reach the tumor tissue, they need to
a. Be internalized by cancer cells,
b. Escape from the endosome into the cytoplasm, and
c. Finally release the siRNA payload to form RISC (Figure 1B).
Kidney glomeruli work as a physical filtration barrier that allows water and small molecules to pass into urine while larger molecules are retained in the circulation [2]. The pore size of the glomerular basement membrane (GBM) is reported to be approximately 6-10nm. Thus, naked siRNAs with a size of approximately 7nm in length and 2 nm in diameter [3], as well as their degraded products, can be readily filtered within 10 min through GBM and passed into urine [4,5]. These facts generate the lower size limitation of approximately 10 nm for design of delivery vehicles. Meanwhile, it is believed that various solid tumors have defective “leaky” vascular structures associated with immature lymphatic ducts, compared with healthy organs/tissues, permitting the accumulation of nanoparticles with several tens to hundred nm in tumor tissues. This size-mediated tumor accumulation mechanism of nanoparticles (or macromolecular drugs) was originally observed by Y. Matsumura and H. Maeda in 1986, and was termed the enhanced permeability and retention (EPR) effect [6]. To date, the size-mediated tumor accumulation of nanoparticles has been widely demonstrated in various tumor-bearing murine models using polymeric micelles, inorganic nanoparticles, and lipid nanoparticles [7-9]. Of importance in this regard is that the tumor accumulation behavior of nanoparticles is significantly affected by the pathophysiology of tumor tissues [10].
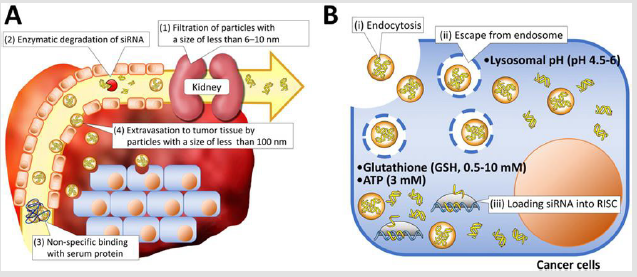
Figure 1: Schematic illustration of delivery barriers in extracellular
A. And intracellular
B. Regions.
In 11 canine cancer patients with spontaneous solid tumors, 110nm-sized nanoparticle displayed high uptake levels in six of seven carcinomas, whereas the same nanoparticle accumulated only one of four sarcomas [11]. Highly permeable tumor models, such as the colon adenocarcinoma LS174T model, are reported to allow significant accumulation of nanoparticles that are even 400nm in diameter [12]. On the other hand, subcutaneous pancreatic BxPC3 tumors are reported to have thick fibrotic stroma and hypo vascularity, hampering the tumor accumulation of N50nm-sized nanoparticles, but not 30 nm-sized ones. Eventually, only 30nmsized nanoparticles accomplished significant antitumor activity in the pancreatic tumor model [13]. These facts have encouraged researchers to engineer smaller delivery vehicles with a size of less than 50nm for enhanced accumulation in heterogeneous tumor tissues. Meanwhile, the EPR effect in human patients has been observed in a handful of examples and is noteworthy [14,15]. A polymer-drug conjugate with a molecular weight of approximately 15kDa, termed SMANCS, which is able to bind to blood albumin and thereby shows significantly increased blood circulation, was found to accumulate in clinical hepatocellular and renal cell carcinomas with a high vascular density through arterial infusion [16]. With a diameter of approximately 90 nm, PEGylated liposomal doxorubicin or Doxil® has a circulation half-life of 21-90h [17-20], exhibiting a 10-fold greater selective accumulation in metastatic breast carcinoma tissue compared to tumor-free skeletal muscle in two patients [21]. The EPR effect observed in animal models needs to be carefully interpreted for the translation to clinical settings as described in other reviews [22-24].
Materials and Methods
Design Criteria to Overcome Extracellular Barriers
Transports of nanoparticles from blood vessels to cancer cells are governed by particle dynamics regarding physical barrier of stroma. The recent observation onto tumor microenvironment showed that tumor blood vessels undergo time-limited formation of an opening in the vessel walls followed by brief outward flow of fluid into the tumor interstitial space (termed ‘eruptions’) probably due to hydrodynamic pressure gradient [25]. Both 30nm-sized and 70nm-sized nanoparticles were erupted into tumor interstitial spaces. The 30nm-sized nanoparticles quickly diffused away but the 70nm-sized nanoparticles was trapped in stroma-rich barriers [25]. Cancer cells mostly surround blood vessels in some clinical tumor (e.g. kidney, brain, liver, thyroid, ovarian, and head and neck cancers), whereas stroma surrounds vessel in other clinical tumor (e.g. breast, pancreatic, colorectal and non-small cell lung cancers) [26,27]. Thus, in case of stroma-rich tumors, nanoparticles need to penetrate into (or distribute across) the stroma tissue to reach the cancer cells nests. In this regard, the penetrability of nanoparticles is reported to significantly depend on their particle size as follows. 20nm-sized PEGylated gold nanoparticles permeated to 40- 50μm depth from vessel centers in subcutaneous MDA-MB-435 tumor, which was deeper than 60 and 100nm-sized nanoparticles [28]. Also, 12 nm-sized PEGylated nanoparticles diffused to approximately 80μm depths from vessel centers in subcutaneous melanoma tumor [29]. These results indicate that smaller nanoparticles are preferred to diffuse into stroma-rich extracellular matrix. On the other hand, rapid proliferation of cancer cells and the resulting blood and lymphatic vessel compression can induce higher interstitial fluid pressure in the tumor area, preventing efficient diffusion of nanoparticles from vessel to cancer cells [30].
Treatment of transforming growth factor-β inhibitor and collagenase can reduce the pericyte coverage of endothelium and fibrosis in the tumor microenvironment, respectively, allowing the enhanced penetration of nanoparticles [31,32]. Of note, the majority of tumor xenograft model in mice relatively lacks stroma, compared to human patients’ tumors. This indicates that the delivery efficacy of nanoparticle is likely over-estimated using inadequate animal tumor models [27]. It is important that the aforementioned size of delivery vehicles must be maintained even in the bloodstream including a huge amount of biomacromolecules and cells. Thus, delivery vehicles should be carefully designed to avoid undesired aggregation and rapid dissociation in the biological milieu until the target tumor region is reached. In particular, positively charged delivery vehicles may electrostatically bind to negatively charged serum proteins and proteoglycans, such as albumin and heparan sulfate, resulting in their aggregation and/or dissociation. Transmission electron microscopy (TEM) has shown that the intravenously injected CALAA-01 has a zeta-potential of 10-30mV and was entrapped with GBMs, which have a high density of heparan sulfate [33], resulting in the loss of their structural integrity. In another study, siRNA-loaded cationic polysaccharide nanoparticles were transferred from the kidney to the bladder more slowly compared with naked siRNA. Considering that the original size of the nanoparticles (220-230nm in diameter) was larger than the pore size of GBMs, the siRNA transfer to the bladder implies that siRNA payloads were gradually released from the nanoparticles and that GBM partially contributes to disassembly of the intravenouslyadministered nanoparticles. Of note, nanoparticles with a size of several hundred nanometers can be engulfed by Kupffer cells through phagocytosis and entrapped by the reticuloendothelial system in the liver and spleen [34-36]. Thus, absorption of serum proteins on to delivery vehicles likely reduces the blood circulation property of delivery vehicles [37]. An effective approach to limit protein absorption is surface-coating with non-ionic, hydrophilic, and flexible polymer brushes, e.g., PEG, poly(N vinyl pyrrolidone) (PVP), poly(N-(2-hydroxypropyl) methacrylamide) (PHPMA), and poly(oxazoline), which all generate steric repulsive forces on the nanoparticle surface [38]. The impact of these polymer brushes on circulation kinetics and biodistribution of delivery vehicles was well explained in other reviews [39-41]. Among these polymer brushes, PEG has been most widely used over many years, and approved as an injectable material by Food and Drug Administration (FDA) in United States [42,43].On the other hand, a few reports on the limitations of PEG, e.g. immunogenicity, have been published after two decades of clinical usage [41,44]. Whereas the immunogenicity of PEG in patient was not reported after treatment with PEGylated liposomes or micelles, PEGylated phenylalanine ammonia lyase tested in Phase I trials induced antibodies against PEG within 6 weeks after single subcutaneous administration [45]. This anti-PEG antibody reduced the efficacy of PEGylated asparaginase because it may eliminate the PEGylated protein from the bloodstream [46,47].
Design Criteria to Overcome Intracellular Barriers
Once target tissues or cancer cells have been reached, delivery vehicles should interact with the cellular surface for internalization into cells. The main parameters that determine endocytosis of delivery vehicles are shape, size, and surface chemistry. These parameters are believed to affect not only the cellular uptake efficiency but also the endocytotic route [48]. The shape effect of delivery vehicles is not described in this review because most delivery vehicles for cancer therapy are constructed to possess a spherical morphology through simple self-assembly procedures or natural growth of seed inorganic particles (see the reference [49,50] on the shape effect). Multimolecular delivery vehicles, e.g., polymeric micelles and LNPs are generally constructed to be 30- 100nmin size, to promote the EPR effect. Inorganic nanoparticles are also reported to demonstrate a size effect on endocytosis; bare gold nanoparticles with 20-50nm in diameter have demonstrated that the most efficient cellular uptake between 10 and 100nm size ranges in cultured cancer cells because gold nanoparticles with these size ranges may balance between the elevated elastic energy associated with increased curvature of the cell membrane and reduced entropy associated with receptor/ligand immobilization [51-53]. Surface chemistries of delivery vehicles are apparently more critical for their endocytosis, compared to size and shape. Positively charged nanoparticles have a high affinity to negatively charged proteoglycans expressed on the surface of most cells, resulting in more efficient adsorptive endocytosis, compared with neutral and negatively charged nanoparticles. Of note, heparin sulfate proteoglycans, comprise transmembrane proteins termed syndecans, are considered major binding sites for cationic delivery vehicles [54]. However, such cationic nanoparticles are not able to take advantage of systemic administration due to nonspecific interactions with negatively charged blood components before reaching target cells. PEGylation of delivery vehicles is a standard strategy, which suppresses such aggregate formation [38]. Nevertheless, PEGylation of delivery vehicles concurrently generates disadvantages for cellular entry due to weakened interactions with the surface of target cells (termed PEG dilemma) [55].
To overcome this PEG dilemma, ligand-mediated targeting strategies have been explored for delivery vehicles to selectively bind to receptor molecules that are overexpressed on targeted cancer cells (other reviews summarize promising ligand candidates, including small molecules, peptides, antibody, and aptamers [56,57]). Arginine-glycine-aspartic acid (RGD) peptide and folate are typical ligands used in siRNA delivery for various types of cancers because these ligands are closely related to angiogenesis of tumor development and metabolism of fast-growing cancer cells. The RGD peptide can strongly and specifically bind to αvβ3 and αvβ5 integrin receptors, which are overexpressed on many cancerous and neovascular endothelial cell surfaces [58,59]. A cyclic form of the RGD peptide (cRGD) provides the rigid structure for enhanced affinity to the target integrins (KD = approximately 40nM for αvβ3 integrin [60]) and prevents degradation of the highly susceptible aspartic acid residue [58]. Folate is a low molecular weight vitamin required by all eukaryotic cells for 1-carbon metabolism and the synthesis of purines and pyrimidines. It has a high affinity (KD = approximately 10nM) for folate receptor isoform α (FR-α), which is highly overexpressed on the surface of ovarian, uterine, brain, and CNS cancers, whereas a high to moderate level of FR-α expression is detected in lung, kidney, and breast cancers [56,61]. Monoclonal antibodies and their fragments are also utilized to recognize specific molecules (i.e., antigens) on the surface of cancer cells. The structure of antibody divides into two different bio functional subdomains. The antigen-binding fragment (Fab) mediates antigen recognition via complementarity-determining regions and the crystallizable fragment (Fc) recruits Fc receptor on the immune cell or the other antibody recognition [62].
Trastuzumab, an antibody for FDA approved antibody-drug
conjugate Trastuzumab emtansine, has KD = 1-7nM for the
transmembrane tyrosine kinase receptor (HER2) [63-65]. Fab
fragments can be used to reduce the bulky size of a full antibody
(approximately 15nm), alleviating immunogenicity and improving
the pharmacokinetic profile of delivery vehicles [61,66]. Nucleic
acid aptamers, which are single-stranded oligonucleotides with
a specific 3D structure, also exert high binding specificity to their
target molecules [61]. To date, no ligand-installed multimolecular
delivery vehicle containing oligonucleotides or small molecular
drug goes to markets [67]. On the other hand, two antibody-drug
conjugates are approved by FDA for treatment of lymphoma and
HER2-positive breast cancer [63]. In subcutaneous folate receptorpositive
tumor mice model, folate-conjugated Vinca alkaloid, EC145,
showed complete cures without a relapse for > 90 days post-tumor
implantation in 4/5 mice [68]. However, in Phase II trials combined
with pegylated liposomal doxorubicin (PLD), median progressionfree
survival (PFS) of EC145 plus PLD marginally increased from
2.7 to 5.0 months compared to PLD-alone control in ovarian
cancer [69]. In recent report, Phase III study was stopped because
EC145 did not demonstrated efficacy regarding PFS in patients.
One plausible explanation is that receptor properties in animal’s
tumor did not represent properties of primary cancer cell found
in a patient’s tumor [23]. Other gaps between laboratory animal
model and human patients in tumor targeting were well explained
in interesting perspectives [23,70].
After endocytosis, siRNA-loaded delivery vehicles encounter a
sequential pH drop in the early endosome (pH 6.5), late endosome
(pH 6.0), and lysosome (pH 4.5-5.0) [71,72]. In this way, the
endocytosed delivery vehicles (and siRNA payloads) are subjected
to lysosomal hydrolysis and inactivated for RNAi machinery. Thus,
delivery vehicles need to contain a functionality to breach the
endosomal membranes for translocation to the cytoplasm. Many
previous studies have demonstrated that polycations containing
low pKa amines and their polyion complexes (PICs) with nucleic
acids can induce endosome disruption (or endosomal escape),
resulting in high transfection efficiency [73,74]. Endosomal escape
induced by low pKa amines has been explained by two possible
mechanisms. One is the proton sponge hypothesis based on
increased osmotic pressure [75,76]. Low pKa amines can protonate
in acidic endosomal compartments and induce proton influx into
endo/lysosomal compartments accompanied by counter ions,
mainly chloride ions. This ion influx increases the osmotic pressure
in endo/lysosomal compartments, possibly eliciting the membrane
destabilization. Of note, the endosomal escape of siRNA elicited
by a huge excess of cationic polymers in cultured cells does not
predict in vivo efficacy in animal experiments because unbound
cationic polymers may not accumulate in the same cancer cells as
delivery vehicles do. In addition, it has gradually been believed that
this hypothesis does not work well. Polycations with high pKa still
induce comparatively high endosomal escape in cultured cells [74].
The other mechanism is direct membrane destabilization by
highly charged polycations [74,77]. As aforementioned, polycations
can bind to the oppositely charged cellular membrane and perturb
membrane integrity. In particular, polycations bearing low pKa
amines can significantly elevate their positive charge density
through amine protonation in endo/lysosomal compartments, and
consequently perturb the endo/lysosomal membrane integrity
for membrane destabilization. The design strategies of delivery
vehicles, which are capable of endosome disruption, will be
described later in this chapter.
Results and Discussion
Design of siRNA Delivery Vehicles
Delivery vehicles have been developed
a. To stabilize their multimolecular structures in the bloodstream;
b. To selectively release siRNA from the stabilized structures into
the cytoplasm,
c. To specifically recognize the target cellular surface, and
d. To allow efficient endosomal escape (Table 1).
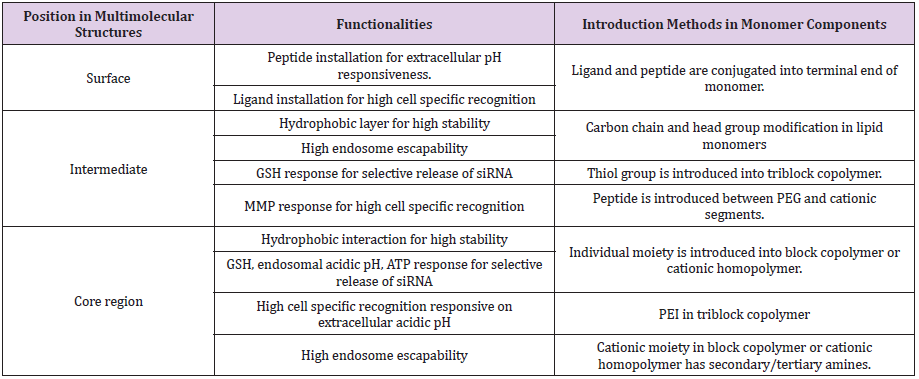
Table 1: Summary of design strategies in this research article.
Various technologies (or strategies) have been applied to mainly elaborate three compartments of multimolecular structures, which are surface, intermediate layer, and core. In this review, multimolecular structures are defined as polymer- and lipid-based nanoparticles. Both nanoparticles comprise cationic monomer components to efficiently encapsulate negatively charged siRNA and increase the siRNA load in nanoparticles. In addition, these components drive electrostatic interaction with siRNA and spontaneously assemble into a multimolecular structure in the aqueous milieu [78,79].
Carrier Design for Stability and Release
Multimolecular delivery vehicles are stabilized by various
driving forces, e.g., electrostatic interaction, hydrophobic
interaction, and hydrogen bonding. Design of building components
to maintain multimolecular structures is a key point to increase
delivery efficacy. Various features in building components, e.g.,
chemical structures and length of building components (cationic
moieties, non-ionic/hydrophilic moieties, and hydrocarbon tail of
lipid) contribute to vehicle stability [80-82]. These features need to
be optimized in the individual delivery vehicles. Among them, pKa
of cationic building components is generally well studied in terms
of vehicle stability and particle formulation. An ionizable cationic
lipid in SNALP contains a dimethylamino headgroup (pKa =6.7±0.1),
efficiently formulating a multimolecular assembly with siRNA at
pH 4. At physiological pH, this vehicle maintains a neutral or low
cationic surface charge density to avoid non-specific disruption
of plasma membranes. Polymeric complex delivery vehicle is also
significantly influenced by pKa of the functional group in cationic
components. The primary amines (−NH2) in polylysine have pKa of
10.5 and are protonated into -NH3+ at 99% in pH 7.4, whereas the
secondary amines in polyethyleneimine (PEI) have pKa of 6.6 and
are protonated at approximately 50% in the neutral pH condition
[77]. Thus, two-fold quantities of secondary amines in PEI are
necessary to neutralize single negatively-charged siRNA compared
with primary amines in polylysine.
Meanwhile, internalized delivery vehicles need to release siRNA
payloads into the cytoplasm for siRNA loading into RISC proteins for
RNAi. The target site-selective release of siRNA can be accomplished
by designing the vehicles that respond to different gradients of
biological signals between intracellular and extracellular regions.
As stabilizing/releasing strategies, multimolecular structures can
dissociate to release encapsulated siRNAs in response to three
representative biological signals: redox potential, pH, and ATP
concentration. The design strategies of multimolecular structure
for these kinds of biosignals are divided into two categories. The
first category is covalent conjugation of siRNA into delivery vehicles
through biosignal-responsive crosslinkers. The terminal end of
siRNA is readily modified with biosignal-responsive chemical
moieties and then associates with the vehicles. These vehicles can
release siRNA through cleavage of the crosslinks in the presence of
higher amounts of biosignals. The other category is construction of
multimolecular assembly using biosignal responsive components.
In this way, the multimolecular structure can dissociate into building
components and simultaneously releases encapsulated siRNAs.
This biosignal responsive disassembly of the multimolecular
structure elicits the rapid siRNA release in the target site.
Hydrophobicity-Stabilized Delivery Vehicles: Stabilization of
delivery vehicles by hydrophobic interaction in aqueous solutions
has been investigated in the early development of vehicles because
of simple chemistry for introduction of hydrophobic moieties
into component materials [83,84]. Hydrophobic moieties, such as
alkyl chains and cholesterol, installed into cationic components
can assist the spontaneous assembly of multimolecular structures
with siRNA through hydrophobic interactions, rendering delivery vehicles more resistant to dissociation (Figure 2A). This increase in
the association number (e.g. the number of building components in
single delivery vehicle) between hydrophobic cationic components
and siRNA results in a higher resistance against serum-containing
media compared with unmodified cationic components [85,86].
Consequently, the hydrophobized delivery vehicles permitted more
efficient cellular uptake of siRNA payloads, leading to enhanced
endogenous gene silencing in cultured cancer cells. However, the
higher stability of delivery vehicles in serum-containing media does
not guarantee the stability in the bloodstream. When hydrophobized
PEG-polycations formulated with siRNA, blood circulation property
increased for only 10 min after tail vein administration [87,88].
The resulting delivery vehicles exerted inefficient tumor growth
inhibition in a subcutaneous model of tumor, indicating that simple
hydrophobic moiety introduction into cationic components is not
enough to generate the stability of vehicles in bloodstream.
In this regard, a previous study demonstrated that vehicle
stability could be further improved by compartmentalizing the
hydrophobic moieties within the multimolecular structure [89]
(Figure 2B). The exclusion of hydrophilic siRNA payload as well
as cationic segments from the hydrophobic core allowed for more
stable assembly because the hydrophobic components were more
tightly packaged in the core without interferences of hydrophilic
siRNAs and polycations. This vehicle had better stability in the
bloodstream compared with the control vehicle without the
compartmentalization of hydrophobic moieties. The intermediate
layer in multimolecular structure can also be stabilized by
hydrophobic moieties (Figure 2C). This type of delivery vehicles
(e.g. mostly lipid-based nanoparticles) contain siRNA in the core
and exhibit longer blood circulation properties compared with the
core-hydrophobized delivery vehicles [90,91]. Cationic lipid-based
core was coated with siRNA and further formulated with anionic
PEGlipid as outer bilayer, utilizing the alkyl chain-constituted
intermediate bilayer stabilized the delivery vehicle [90]. This
delivery vehicle (a particle size of 100nm) has a plasma half-life
of approximately 18h and tumor accumulation of siRNA peaked at
24h after systemic administration.
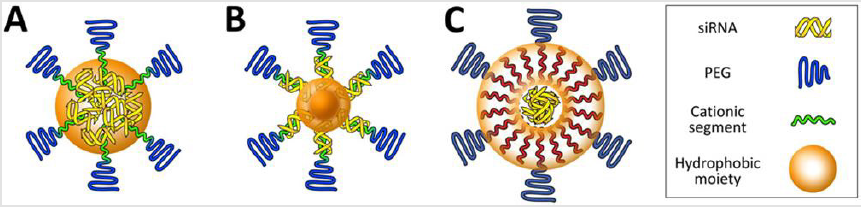
Figure 2: Platform structures for the design of delivery vehicles with high stabilies. Multimolecular structures have hydrophobized core (A, B) or intermediate layer (C)..
Furthermore, significant fluorescence of fluorescent dyelabeled siRNA could be detected in the tumor tissue until 72h after administration. Another delivery vehicle is composed of cationic lipid, azide modified cholesterol, and anionic alkyne-modified hyaluronic acid (HA) [92]. Cationic lipid nanoparticle contains siRNA and azide modified cholesterol in the core and intermediate layer, respectively. Interestingly, the anionic alkyne-modified HA was covalently conjugated to the azide-modified cholesterol, reducing interference of integrity of the intermediate bilayer. The delivery vehicle had a particle size of 130nm and exhibited a plasma halflife of 3-4h. The vehicle showed better tumor growth inhibition, compared to controls e.g. lipid nanoparticle electrostatically-coated with HA and cationic lipid nanoparticle without HA. This result probably indicates that the integrity of hydrophobic intermediate layer affects stability of multimolecular structures. However, these delivery vehicles with longer blood circulation properties exhibited dose ranges of siRNA between 1.2mg/kg and 7.5mg/kg for tumor growth inhibition in animal models. These values are similar to those with other vehicles with shorter blood circulation properties, implying that selective release of siRNA and better endosomal escape ability may be further required for reducing the dose amount of siRNA and increasing the gene silencing efficiency.
Delivery Carrier Design for Selective Release of siRNA
Redox Potential Responsive Delivery Vehicles: Glutathione
is a thiol containing tripeptide composed of glutamic acid (Glu),
cysteine (Cys), and glycine (Gly), and works as a main antioxidant
in cells. Glutathione is distinguished from other common peptides
by the unique structure of γ-glutamate, rendering glutathione inert
to normal intracellular peptidase-mediated degradation [93]. The
cell maintains reduced glutathione (GSH) by de novo synthesis
from the three amino acids and reduction of oxidized glutathione
(GSSG) by glutathione reductase, which induces the concentration
of GSH to be 50-1000 times higher than that of GSSG in cells [94,95].
In this way, GSH concentrations can be distinguished between the
intracellular and extracellular environment. GSH concentration
within cells is 0.5-10mM but decreases to 10-30μM in blood plasma
[93-95]. Thus, the disulfide linkage can be preferably cleaved in the cytoplasm or intracellular compartments while it is slowly
degraded during blood circulation. The payload drug release from
disulfide crosslinked nanoparticles was observed from 2-4h after
internalization by the cells in cultured cells [96]. Thiol functionality
greatly contributes to reversible stability of delivery vehicles when
disulfide bonds are introduced into the intermediate layer or the
core.
Multimolecular structure maintains (or slowly dissociate) its
pre-formed structure in bloodstream, facilitates tumor accumulation
by EPR effects or ligand-mediated targeting, disassembles in the
GSH-rich cell interior, and eventually deliver the amount of siRNA
in RISC to induce desired RNAi. As a representative, the primary
amine group in the side chain of PEG-polycations was modified
with 2- iminothiolane (2IT) to introduce a free thiol group as well
as an amine group [97]. The resulting disulfide crosslinked delivery
vehicle had a thiolate core structure with 40-50nmdiameter
size and neutrally charged with zeta potential of 0.1mV (Figure
3A). The vehicle improved blood circulation property of halflife
(approximately 10min) compared to naked siRNA and noncrosslinked
vehicle control (both half-lives 3-4min). The improved
blood circulation affected the biodistribution of fluorescent dyelabeled
siRNA in animal experiment. The similar amount of siRNA
was accumulated in kidney administered by naked siRNA and the
non-crosslinked vehicle at 24h after injection of vehicles, whereas
half of the siRNA amount was observed in kidney administered by
the crosslinked vehicle.
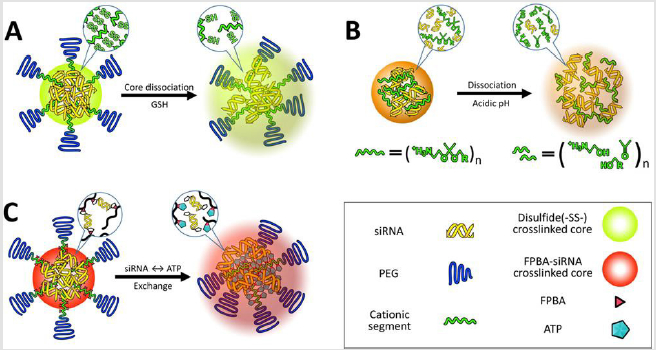
Figure 3: Illustration of delivery vehicles for selective release of siRNA. Multimolecular structures introduce redox-responsive thiol bonds in the core region
A. Delivery vehicle contains acidic pH responsiveness for faster dissociation
B. And introduce ATP-responsiveness in the core region for selective dissociation (C).
Moreover, a crosslinked vehicle with ligand installation delivered approximately two-fold higher amount of siRNA in tumor compared to the controls (e.g. naked siRNA, a non-crosslinked vehicle, and a crosslinked vehicles without ligand installation). Finally, this crosslinked vehicle improved tumor growth inhibition in subcutaneous cervical tumor (25μg siRNA/mice, 6 injections), demonstrating that the selective release of siRNA in tumor was successfully accomplished. Stability of disulfide crosslinked delivery vehicles can be further increased when hydrophobic moieties are simultaneously formulated in the core through additional hydrophobic interaction. In this term, ligand-installed and thiolated PEG-polycations were formulated with cholesterol-modified siRNA (Chol-siRNA) [98]. The resulting delivery vehicles were stabilized by additional cholesterol mediated hydrophobic interactions as well as by thiol crosslinking. The vehicle increased blood circulation properties with a half-life ≤ 20min compared to naked siRNA/ ligand-installed and thiolated PEGpolycations control, leading to higher tumor accumulation of siRNA and enhanced RNAi efficacy in subcutaneous cervical tumor. These researches clearly showed that increased stability/selective release of siRNA can achieve higher RNAi efficacy in an animal tumor model. Thiol crosslinking strategy in the core of multimolecular structure needs to consider an innate phenomenon of preferable intermolecular disulfide linkages. When large quantities of the thiol group are introduced into the core region or intermediate layer, the possibility of intramolecular disulfide linkage is higher than that of intermolecular disulfide linkage. Thus, the current disulfide crosslinked nanoparticles showed restricted redox responsive stability, indicating that the increase in intermolecular disulfide crosslinking will be a key factor to improve RNAi efficacy of delivery vehicles in passive or ligandmediated targeting.
Disulfide linkages are introduced into a clinically ongoing polymer/siRNA conjugate vehicle (named Dynamic Poly Conjugate) for systemic delivery of siRNA into viral infected hepatocytes [99]. This conjugate vehicle consists of an endosomolytic backbone polymer conjugated with thiolated siRNA, N-acetylgalactosamine as a targeting ligand and PEG. In a Phase I clinical trial in healthy volunteers, a single dose of Dynamic Poly Conjugates was well tolerated up to 2mg/kg when administered intravenously [100]. Adverse events were reported to be mild or moderate. Serum hepatitis B surface antigen was reduced by up to 50% after a single dose of 2mg/kg and significant reductions were detected between 43 and 57 days in a Phase IIa trial.
Acidic pH Responsive Delivery Vehicles
Acidic pH (pH 4.5-6) in late endosomes has frequently been highlighted as a representative biosignal for triggering the sitespecific drug release from delivery vehicles because a large pH change from extracellular neutral pH to endosomal acidic pH allows us to utilize acid-labile chemistry [101]. Various acid-labile bonds, such as acetals [102], hydrazones [103], β-thiopropionate [104], phosphoramidate [105], orthoesters [106], and citraconic amide [107,108], have been applied to construct multimolecular structures that elicit acidic pH-responsive release of drug payloads, including anticancer drugs and biomacromolecules (Figure 4). For instance, amino ketal linkages were installed into a polycation backbone and the resulting ketalized polycations formulated siRNA into nanoparticles, which is highly charged with zeta potential of 16 to 22mV (Figure 3B) [109]. The ketalized vehicles efficiently disassembled and released more quantities of siRNA into the cytoplasm whereas unmodified control vehicles exhibited delayed dissociation and captured siRNA at 4h of incubation in cultured cells. Furthermore, the unmodified control vehicles (and loaded siRNA) were observed both in the cytoplasm and the nucleus because their high surface charge probably induced non-specific interaction with membrane of the nucleus. This ketalized delivery vehicle improved RNAi efficiency in cultured cell condition and clearly showed advantages of selective siRNA release. In other example, siRNA is conjugated with a maleic acid derivative amide into anionic polymer and the resulting anionic polymer/siRNA was further formulated with cationic polymers [110]. Released siRNA from the vehicle was not detected at pH 7.4 in 1h incubation whereas approximately 30% of released siRNA was observed at pH 5.0. However, RNAi efficiency of the vehicle was moderately improved in cultured cancer cells compared with a pH-unresponsive control. One research reported that cholesterol was conjugated with acetal linkage into PEGpoly(vinyl alcohol) (termed Chol-PVA-PEG) [111]. This Chol-PVA-PEG was further formulated with siRNA and cationic cyclodextrin, rendering a nanoparticle in the size of 120-170nm. Cholesterol moiety facilitated the compaction of siRNA into the nanoparticle through hydrophobic interaction. The acetal linkage of the cholesterol moiety was degraded in late endosome, promoting decondensation of the nanoparticle and release of siRNA. The size of the nanoparticle was not changed at pH 7.4 for up to 24h but the polydispersity of the nanoparticle started to increase after 4 h at pH 5.5. The nanoparticle showed similar RNAi efficiency to PEI and lipofectamine 2000 in cultured cancer cells. Acid-labile chemistry has also frequently applied to the responses of other components in multimolecular structures for delivery of biomacromolecules e.g. detachment of PEG [112], ligand [99], and cationic polymer [113].
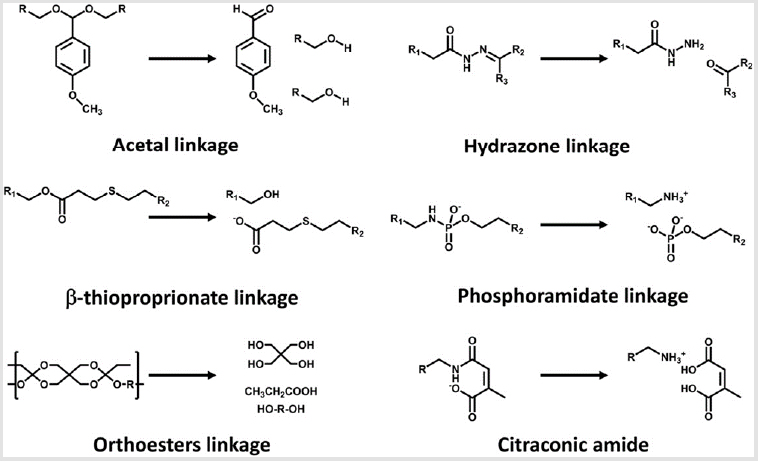
Figure 4: Cleavage of acidic pH-sensitive linkages.
ATP Concentration-Responsive Delivery Vehicles
ATP, the most abundant ribonucleotide, is a responsive
signal for cell-specific release of siRNA. Whereas ATP is present
in the extracellular environment at approximately 0.4mM,
its concentration is dramatically higher up to 3mM within
the intracellular matrix [114]. Similar to the aforementioned
environment-responsive delivery vehicles, delivery vehicles can be
designed to release payload siRNA into the ATP-abundant cytosol
by utilizing phenylboronic acid (PBA) chemistry. PBA can form
reversible covalent esters with 1,2-cis-diols on a ribose ring, which
is present at the 3′end of siRNA and ATP [115]. As a representative,
3-fluoro-4-carboxyphenylboronic acid (FPBA) was conjugated into
primary amino groups in the side chain in PEG-polycations and
complexed with the ribose-terminated siRNA as an intermolecular
crosslinker (Figure 3C). This delivery vehicle was stabilized by
the covalent conjugation between siRNA ribose and ionized FPBA
moiety, hydrophobic interaction derived from non-ionized FPB
Amoieties, and ion-pair formation between siRNA phosphates and
residual amino groups in the polycation. Consequently, the delivery
vehicle containing the ribose-terminated siRNA showed better
stability against counter polyanion exchange with dextran sulfate,
compared to control containing deoxyribose-terminated siRNA at
either or both ends.
On the other hand, this delivery vehicle dissociated in the ATP
concentration range of more than 1mM. Compared to disulfide
crosslinking, this crosslinking further needs to be clarified in terms
of
a. Difference of on-off responsiveness between ATP concentration
and GSH concentration, and
b. Although the thiol group does not affect initial electrostatic
association between the polycations and siRNA, the relatively
hydrophobic and bulky PBA group may prevent efficient
crosslinking between diol and PBA. More precise design of
block copolymer can improve delivery vehicle performance.
The payload release dependent on the gradient of ATP
concentration was also attained by using an ATP-binding
aptamer-incorporated DNA motif for anticancer drug
delivery [116,117]. The Gu group designed a doxorubicinincorporated
aptamer/single stranded DNA duplex, where
the aptamer changed its tertiary structure in the presence of
ATP and lost binding affinity for doxorubicin. The doxorubicinincorporated
aptamer/DNA duplex forms a nanogel with
additions of protamine and hyaluronic acid. Finally, the
nanogels successfully obtained moderate ATP-responsive
growth inhibition in subcutaneous breast tumors.
Delivery Carrier Design for High Cell Specific Recognition
Delivery vehicles enter the tumor microenvironment from neighboring blood vessels. Multimolecular structures that are stable enough in the bloodstream can arrive at the tumor tissue from the leaky vessels. As described above, the vehicle can be further elaborated to possess higher selectivity to individual cancer cells or have more efficient uptake into cells. The tumor microenvironment is composed of many types of cells including malignant cancer cells, fibroblasts, immune cells, and endothelial cells [118]. The delivery vehicle should be designed to recognize its target cells to increase the delivery efficiency. This can be accomplished by ligand-receptor interaction (or active targeting). For efficient recognition, cellspecific ligands are installed onto the surface of delivery vehicles or the distal ends of neutral and hydrophilic spacers. Alternatively, the delivery vehicle is designed to selectively expose the positive charges near target cells, facilitating the binding to the cellular surface. The vehicle detaches neutral and hydrophilic layers or alters its charge to positive charge in response to tumor specific biosignals. The tumor environment-specific biosignals include acidic pH and specific enzymes, which trigger the chemical reaction in delivery vehicles to alter their multimolecular structures.
Biological Stimuli-Responsive Delivery Vehicles: The lowered pH (pH 6.5-7.0) in the deep tumor tissue has gradually paid attentions as acidic pH signals [119]. This slight pH drop in the tumor extracellular matrix originates from the high metabolic rate of cancer cells in poorly perfused regions. Inadequate oxygen delivery to some regions of tumors generates hypoxic condition, which restricts oxidative phosphorylation of pyruvate in the mitochondria. Hypoxic cancer cells shift their glycolysis metabolism from generation of pyruvate to lactate. Increased oncogenic metabolism also generates an excess of protons and carbon dioxide, which are kept in equilibrium with carbonic acid by the enzyme carbonic anhydrase. These weak acids and protons are exported out of the cells, inducing enhanced acidification of the extracellular milieu [119]. The protonation degree of amino groups in cationic components of the delivery vehicle is a key for utilization of the lowered tumoral pH as a biosignal. In this regard, the amine pKa value of cationic components has been highlighted as an indicator for their protonation behavior and regulated to convert the surface charge of multimolecular structures from negative to positive in response to the acidic pH in deep tumor tissues. This strategy expects that higher cellular uptake of positively charged vehicles occurs in tumors, whereas negatively charged vehicles suppress non-specific cellular uptake in healthy tissues. For instance, the delivery vehicle was prepared from branched PEI, which increased a protonation degree when the solution pH decreased from 7.4 to 6.8 [120] (Figure 5A).
The mixing ratios between cationic PEI and anionic siRNA were carefully selected in gel retardation assays to formulate multimolecular vehicles that had negative surface charges at pH 7.4 and positive surface charges at pH 6.8, associated with the facilitated protonation of PEI. The resulting surface charge-reversible vehicles had higher tumor accumulation of fluorescent dye-labeled siRNA than non-charge-reversible negative and positive control vehicles. A similar surface charge conversional vehicle was also developed using peptides comprising histidine and glutamic acid residues [121] (Figure 5B). Considering that the pKa of histidine is approximately 6.0, glutamic acid residues were utilized as neighbors of histidine in the peptide sequence for increasing the pKa value of the basic amino acid via stabilization of the protonated form. Delivery vehicles integrated with this peptide exhibited an increase in surface charge at pH 6.5, compared to pH 7.4, and enhanced the cellular uptake in cultured cancer cells. These studies fully utilized an advantage of multimolecular assembly by amplifying a subtle change in the protonation degree of component amino groups for a dramatic change in surface charge of the assembly
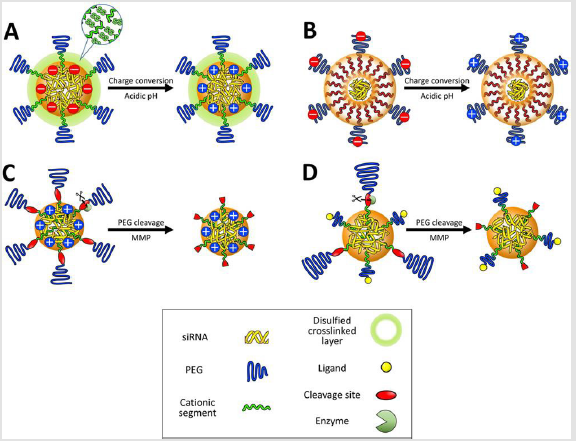
Figure 5:
A. Illustration of delivery vehicles for cell specific recognition in tumor micro environment. Multimolecular structures contain
charge-conversion moieties in the core
B. Or on to the surface
C. Enzymes (e.g. MMPs) cleave PEG layers, which exposes positive charges in the core
D. Or cell-specific ligands.
Alternative strategy for high cellular internalization is that
neutral and hydrophilic shielding layer is torn off in the extracellular
matrix near the tumor whereas it protects delivery vehicle in the
bloodstream. Increased surface charges or exposure of a ligand
moiety induces higher cellular uptake of delivery vehicles [122].
Acidic pH and specific enzymes in the extracellular matrix can
trigger the detachment of the shielding layer. The detachable
strategy has widely been used for delivery of biomacromolecules
such as plasmid DNA and antisense oligonucleotide but more
researches regarding enzyme response than acidic pH response
have been investigated for systemic delivery of siRNA. The vehicle
responding to the subtle pH difference between extracellular
matrix and neutral pH may not have significantly increased
RNAi efficiency compared with the vehicle responding to the pH
difference between late endosome and neutral pH. The invasive
nature of malignant tumors has long been associated with the
ability to degrade collagen in the extracellular matrix, a threedimensional
non-cellular structure that is present in all tissues and
provides physical support for tissue integrity and elasticity [123].
The matrix metalloproteinase (MMP) family is involved in tissue
invasion, angiogenesis, regulation of inflammation, and formation
of metastatic niche [124,125]. MMPs include 23 zinc-dependent
endopeptidases and have been overexpressed from tumor cells
or stromal cells infiltrating the tumor. Therefore, MMPs with a
higher concentration in the tumor environment can also represent
biosignals, which can be used to design intelligent vehicles.
MMP-cleavable peptide (VPLSLYSGCG) is placed between PEG
and the polycations [126] (Figure 5C). When delivery vehicle was
treated with 50nM MMP-7, a similar concentration in the metastatic
tumor micro environment, the zeta-potential of the delivery vehicle
gradually increased over 6.5h and MMP-7 efficiently cleaved PEG
layers. Using MMP-7 treatment at 1-5nM concentrations relevant
to normal and healthy tissue, the vehicle showed a slower rate of
increase in zetapotentials, indicating a dose-dependent response to MMP-7 concentration. MMP7-pre-treated vehicle was internalized
at 2.5-fold higher amounts in cultured cancer cells than non-treated
controls due to the increase in its surface charges. In luciferase
overexpressing cancer cells, the MMP7-pre-treated vehicle also
showed better endogenous luciferase gene silencing than nontreated
controls, implying that PEG-peptide polycation has the
potential to function in an animal tumor model. It is notable that
VPLSLYSGCG peptide can also be efficiently cleaved by other
enzymes, MMP-2 and MMP-9. This PEG-peptide-polycation was
further improved with a folate-conjugated polymer to increase
cellular uptake in the tumor microenvironment [127]. PEG20Kpeptide-
polycation and folate-PEG2K-polycationwere synthesized
and micellar nanoparticles were prepared using a 1:1 mixing ratio
of each polymers. When the micellar nanoparticle arrived at the
MMP-rich environment, the longer PEG shield was cleaved by MMP-
7 and the underlying folate ligand exposed (Figure 5D). Thus, the
nanoparticles can internalize into target cells by ligand-mediated
endocytosis. These nanoparticles achieved greater than 50%
protein-level knockdown in cultured folate receptor expressing
breast cancer cells. MMP responsive delivery vehicle was further
evaluated in an animal model [122].
A PEG-sheddable delivery vehicle through MMP2-cleavable
peptide (PLGLAGR9) significantly enhanced growth inhibition in
subcutaneous breast tumor (20μg siRNA/mice, 7-8 injections).
However, moderate inhibition of tumor growth compared with
controls indicated that MMP of large molecular weight cannot
easily access target peptide in the intermediate layer of the delivery
vehicle, probably inducing a delayed responsiveness in the tumor
environment. High dense PEG layer in the current delivery vehicles
probably guarantees reduction of non-specific absorption of serum
proteins in the bloodstream but simultaneously reduces the MMP
approaches. Thus, biosignals of large Mw such as MMP may not
fully induce desired responses in multimolecular structures. Other
MMP-cleavable peptides and their applicable delivery vehicles are
summarized in (Table 2).
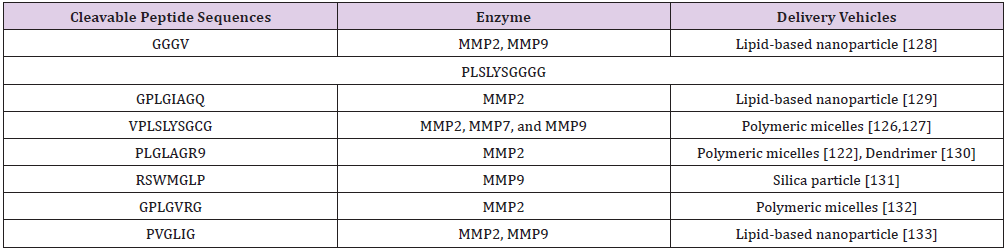
Table 2: List of MMP cleavable peptide sequences and delivery vehicles..
Ligand Installed Delivery Vehicles: To optimize the ligandmediated active targeting functionality, several parameters, including ligand density and length/density of spacer, should be considered for construction of actively targeted multimolecular structures. The underlying mechanism of active targeting is the recognition of the ligand by its target receptors, and thus, a higher density of both ligand and receptor generally guarantee higher opportunities of their binding [128-133]. For example, conjugates of chemically modified siRNA and tri-N-acetylgalactosamine (GalNAc) resulted in higher cellular uptake in primary mouse hepatocytes than bi-GalNAc siRNA conjugates [134]. This result demonstrates a multivalent binding effect of ligands for the enhanced cellular uptake efficiency, at least in cultured cells. Similarly, a higher number of ligands on multimolecular vehicle surface exhibit more efficient internalization in cultured cells. However, such enhanced uptakes through the binding of the multivalent ligands were not always observed for active targeting in systemic administration. The receptors are expressed not only on target cellular surface but also non-target ones at lower levels. Thus, a higher number of ligands can generate the higher affinity (or avidity) to target cellular surface, but concurrently increase the risk for non-specific binding to such non-targeted cells. Intercellular adhesion molecule-1 (ICAM-1) is constitutively expressed at a basal level on endothelial cells in quiescent vasculature but its expression is markedly elevated in pathologically activated endothelium. Introduction of reduced quantity of ICAM-1 specific antibody onto a particle surface enhanced the selectivity for binding to inflamed vasculature compared with normal tissues [135]. The length of spacer between ligand and a nanoparticle surface affects the binding chances due to flexibility of the spacer. Antibody-installed nanoparticles equipped with PEG2000 or PEG3000 exhibited greater binding to cultured dendritic cells, compared to those with PEG6000, PEG10000, or PEG20000 [136]. The optimal PEG length depends on individual delivery vehicle. A peptide ligand-installed liposome with PEG350 linker dramatically enhanced cellular uptake in cultured cancer cells with appropriate density of the peptide, whereas control liposome with PEG2000 linker showed the similar level of cellular uptake to nontargeted controls [137].
The density of PEG onto nanoparticles also affects cellular uptake amount by active targeting. cRGD installed nanoparticle tailored with 5% PEG2000 density exhibited the highest relative internalization amounts in cultured cancer cells and animal experiments compared to no ligand-installed control with the same PEG density [138]. Other nanoparticles tailored with 10%- 50% PEG2000 densities showed less active targeting effects. There are other factors such as charges of ligand/delivery vehicle and size of delivery vehicle to obtain efficient active targeting and these are precisely described in other researches [56,139]. Interestingly, delivery vehicles can utilize blood components for active targeting. Cholesterol-conjugated siRNA with a partial phosphorothioate backbone and 2′-O-methyl-modified nucleotides binds to lowdensity lipoprotein (LDL) (KD = 100μM) and obtained a plasma half-life of approximately 100-120min (dose amount: 50mg/kg), accompanied by significant gene silencing in the liver through LDL receptor-mediated endocytosis [140,141]. Some lipid-based nanoparticles are also believed to exchange their components with serum and adsorb lipoproteins, leading to enhanced internalization into hepatocytes through lipoprotein receptors [142].
Delivery Vehicles for High Endosomal Escapability
Delivery vehicles potentially contain endosomal escapability and facilitate their escape from endo/lysosomal acidification. In cultured cells, this enhanced endosomal escapability of vehicles greatly increases gene silencing efficiency compared with controls without escapability. In systemic administration, the functional chemical groups of endosomal escapability, e.g., secondary/tertiary amines or histidine should be carefully tailored in multimolecular structures because those possess a low pKa and unprotonated amino groups do not participate in electrostatic association with siRNA. PEI is a representative cationic polymer eliciting endosomal escapability through the proton sponge hypothesis [75,76]. PEI shows partial protonation of nitrogens at physiological pH (45% in linear PEIDP520) and augmented protonation at endo/lysosomal acidification (55% in linear PEIDP520) in 150mM NaCl [77,143]. One notable disadvantage of PEI is cytotoxicity. It is known that the cytotoxicity is substantially elevated with an increase in molecular weight of PEI. However, low molecular weight PEI cannot maintain stable multimolecular structure under physiological milieu because of its less ion pairing sites to nucleic acids, compromising the transfection efficacy. Thus, the low molecular weight PEI (e.g. molecular weight 800Da) was conjugated with each other to have a higher molecular weight (e.g. average molecular weight 10-20 kDa) through biodegradable linkages for maintaining higher transfection efficiency associated with lower cytotoxicity [144,145].
A low toxic and pH-dependent cationic moiety was alternately developed by fine-tuning the number of repeating amino ethylene unit, - (CH2CH2NH)n- in the side chain of polyaspartamide [74]. The polymers were synthesized by direct aminolysis of PEG-b-poly(β-benzyl-L-aspartate) with diethylenetriamine [-(CH2CH2NH)2-, DET] or tetraethylenepentamine [-(CH2CH2NH)4-, TEP] [termed as PEGPAsp(DET) and PEG-PAsp(TEP), respectively] [146]. PAsp(DET) contained 51% and 82% protonated amino groups at pH 7.4 and pH 5.5, respectively, thereby eliciting a large change in protonation degree (Δα =31%) [77]. PAsp(DET) had a monoprotonated structure (-CH2CH2NH-CH2CH2NH3+) to induce a low membrane destabilization effect at pH 7.4, whereas the diprotonated structure of PAsp(DET) (-CH2CH2NH2+-CH2CH2NH3+) had a high membrane destabilization effect at pH 5.5. PAsp(TEP) also exhibited acidic pH-selective membrane destabilization ability, which is only possessed by even numbered aminoethylene units: PAsp(DET) and PAsp(TEP) had two and four aminoethylene units, respectively. Further detailed and comprehensive mechanisms regarding the endosomal escape moiety are explained in other review papers [77]. Derivatives of these polymers, PEGPAsp(DET) and PEG-PAsp(TEP), successfully delivered therapeutic siRNA into subcutaneous and spontaneous model of cancers [87,88,147].
Other chemical structures in cationic polymer also facilitated pH mediated membrane disruption. For example, poly (dimethyl aminoethyl methacrylate-co-propyl acrylic acid-co-butyl methacrylate) [termed as p(DMAEMA-co-PAA-co-BMA)] was ampholytic under physiological pH in that positive DMAEMA and negative PAA residues masked hydrophobic BMA [148]. At endosomal pH, PAA underwent a hydrophilic-to hydrophobic transition of carboxylate groups and DMAEMA (pKa = 7.4) increased the positive charge in the residues. This changed the polymer from a hydrophilic polyampholyte to hydrophobic polycations that were capable of disrupting the endosomal membrane. In addition, the imidazole ring (pKa=6.0) of histidine induces a proton sponge effect in an acidic condition and poly(histidine) has been utilized as an endosomal escape moiety [149]. Although some cationic lipids were synthesized by linking the alkyl chain or lipid components with PEI to provide buffering effect [150,151], the lipid-based nanoparticle had its own endosomal escape mechanism, which was first proposed by the Szoka group [152,153]. Cationic components of lipid-based nanoparticles are associated with anionic phospholipid in the endosomal membranes, promoting the formation of the inverted hexagonal (HII) phase. The HII phase involves six cylindrical fused structures of two oppositely charged lipid components, where the head group of the lipid components faces inward and the hydrocarbon tail faces outward. Cationic lipids promoting these non-bilayer structures lead to disruption of the endosomal membrane and release of siRNA from nanoparticles into the cytoplasm [152,154]. Dioleoyl phosphatidyl ethanolamine (DOPE) is known as a “fusogenic” cationic lipid because it can place this non bilayer structure, whereas dioleylphosphatidylcoline (DOPC) forms a stable lamellar structure that dramatically decreases lipid-based nanoparticle fusion with the endosomal membrane.
Delivery Carrier Design in Other Category
Layer-by-layer delivery vehicle: Delivery vehicles constructed by layer-by-layer (LbL) technology for high loading of siRNA has been an attractive strategy for local administration because the vehicle has superior gene silencing efficiency even at picomolar siRNA concentrations in cultured cells [155,156]. Nevertheless, its larger particle size (> 100 nm) and wide size distribution of the particle have been believed to be drawbacks for passive/active targeting by systemic route. Very recently, a systemically injected layer-by-layer (LbL) particle was developed to exhibit comparative tumor growth inhibition in animal models when the particle was carefully engineered to possess building components for tumor targeting (Figure 6A). LbL nanoparticle construction, which alternately deposits siRNA and polycations on to template, has a unique advantage over other multimolecular structures because a single nanoparticle can load much larger amounts of siRNAs (approximately 3500 siRNA molecules) and exhibit a long period of siRNA release time (approximately 3 weeks) in cultured cells [157]. When an LbL nanoparticle codelivered multidrug resistance protein 1 (MRP1) siRNA with doxorubicin into a subcutaneous animal model of triple-negative breast cancer, MDA-MB-468 cancer, it showed the synergistic inhibition of tumor growth (1mg/kg siRNA and 1mg/kg doxorubicin) [158]. Of note, MRP1 is a cellsurface efflux pump involved in redox regulation of multidrug resistance by clearing the intracellular concentration of xenobiotics and toxins [159,160].
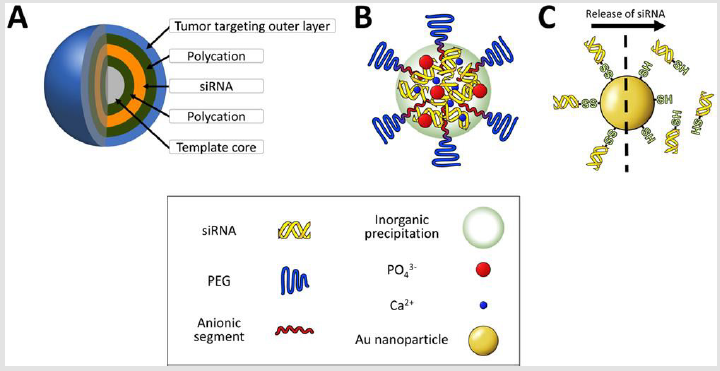
Figure 6: Illustration of delivery vehicles for other categories. An LbL particle is constructed by alternative deposition of anionic and cationic components
A. A calcium phosphate particle stably formulates siRNA and PEG-polyanions in the core region
B. Thiolated siRNA is attached to Au nanoparticles and released in
C. The response of GSH concentration.
Because MRP1 siRNA treatment did not show any tumor inhibitory effects, this result demonstrated that siRNA as supplements can be applied for combination therapy with clinically approved anticancer drugs. At present, to reduce cancer drug resistance, two combination therapies regarding ERCC1 siRNA/cisplatin and mutated KRAS siRNA/gemcitabine are being conducted in Phase I and Phase II studies, respectively. Design strategies of delivery vehicles to increase the efficiency of combination therapy and the targeting gene/cancer drug combination are well described in a previous review [161]. The LbL nanoparticle provides clues in design of delivery vehicles. High loading of siRNA in the single delivery vehicle is also a critical factor for efficient gene silencing. Another point to consider is that altering the size of LbL nanoparticles loaded with high amount of siRNA will increase performance of the delivery vehicles.
Calcium Phosphate-Formulated Delivery Vehicles:
Deposition and co-precipitation of inorganic materials on
multimolecular structures can facilitate to formulate siRNA [162].
These inorganic material-stabilized vehicles have been developed
for better size control of the particles and higher encapsulation of
siRNA. Calcium phosphate (CaP) is one of the most commonly used
component materials because CaP is a mineral of human bone and
generally considered to be biocompatible. CaP precipitates were
used as transfection reagents of plasmid DNA because they can bind
and encapsulate polyanions/ nucleic acids and protect the nucleic
acid from enzymatic degradation. The major limitation of CaP
precipitates was the uncontrollable and rapid growth of CaP crystals
after preparation, resulting in the formation of micrometer-sized
agglomerates. Size-controlled nanoparticles with high colloidal
stability were obtained when CaP was precipitated with a mixture of
PEG-polyanions and siRNA [163]. CaP precipitation simultaneously
formulates phosphate of siRNA and the carboxyl group of the PEG
polyanions (Figure 6B). Precipitated nanoparticles carefully need
to be examined so that single particles encapsulate both building components because there is a possibility that one component
is favored in CaP precipitates. The resulting nanoparticle can
dissolve under highly dilute conditions for payload release, based
on the equilibrium shift toward calcium and phosphate ions. The
resulting CaP nanoparticle has approximately 40 nm diameter size
and maintained its initial size in serum containing medium while
the nanoparticle rapidly dissociated in medium mimicking the
cytoplasm [147,164]. CaP nanoparticles showed better endogenous
VEGF mRNA silencing in cultured pancreatic cancer cells and did
not induce significant toxicity at concentrations of up to 25-fold
higher concentrations than RNAi-induced concentrations.
A high dose of unbound calcium ions can apparently affect the
heart because cardiac excitation-contraction is based on regulation
of intracellular calcium ion concentration in heart muscle cells
[165]. Thus, for systemic delivery of therapeutic siRNA, the CaP
nanoparticle was further purified by an ultrafiltration method and
approximately 80% of the original calcium contents were removed
[147]. VEGF siRNA delivered by the purified CaP nanoparticles
(25μg siRNA/mice, four injections) inhibited subcutaneous tumor
growth with negligible acute toxicity, indirectly indicating that
CaP nanoparticles are stable enough in the bloodstream to attain
gene silencing in remote tumors. While CaP precipitates contribute
to increased stability of block copolymer based nanoparticles,
CaP precipitates can enhance endosomal escape ability. Delivery
vehicles containing CaP precipitates disassemble at low pH in the
endosome, which cause endosome swelling and eruption, releasing
the entrapped siRNA due to a high concentration of ions [166]. This
nanoparticle has been co-formulated with three different siRNAs
against MDM2, c-myc, and VEGF at the weight ratio of 1:1:1 and
significantly reduced lung metastases of B16F10 at a relatively low
dose (0.36 mg/kg siRNA, four injections) [167]. Delivery efficiency
of the current delivery vehicles stabilized with inorganic materials
may increase with more precise design of vehicle components.
When inorganic materials/siRNA are further encapsulated within
the hydrophobic intermediate layer in the core, this structure may
prevent rapid dissociation in the bloodstream.
Gold Nanoparticle-Templated Delivery Vehicles: Gold nanoparticle (AuNP) is widely selected for siRNA delivery because the bonding of gold-thiol group exhibits GSH concentration responsive cleavage. Thus thiolated building components (e.g. siRNA or polymers)-conjugated AuNP is also relatively stable under extracellular condition but these thiolated components can be competed off the AuNP in cell interior. In this term, siRNA is designed to be released from the delivery vehicle in response to GSH concentration (Figure 6C). Thiol-terminated RNA duplexes are loaded into 13nmAuNP and further treated with oligoethylene glycol-thiol or PEG-thiol, as an additional surface passivating ligand [168]. The resulting spherical nucleic acid (SNA) nanoparticles contained approximately 90 sense strand and approximately 38 antisense strands per AuNP. Pharmacokinetic analyses using a two compartment model showed that the blood circulation properties of the SNA nanoparticles had a half-life of approximately 1min in the first phase and approximately 8.5h half-life in the second phase. This data suggests that thiol-gold coordination is stable enough to maintain thiol-terminated RNA duplexes in the bloodstream. The SNA nanoparticle successfully penetrates the blood-brain barrier (BBB) and blood-tumor barrier (BTB), and reduced Bcl2L12 expression in intracerebral glioblastoma multiforme. The reduction in Bcl2L12 mRNA and the subsequent protein level compared with controls indicates that the thiol-terminated RNA duplexes can be efficiently released in GSH-rich cytoplasm and exert appropriate gene silencing. Other example is that thiol-terminated PEGpolycations was complexed with siRNA and then the resulting thiolated complex was conjugated into 20nm AuNP [169]. This delivery vehicle had 40nm diameter size and was loaded with approximately 20 siRNAs per a particle. The blood circulation property of this vehicle showed that 10% amount of the initial dose continued to circulate at 3h and higher tumor accumulation of siRNA was obtained in subcutaneous cervical tumor compared to controls e.g. naked siRNA, thiolated complex, and non-thiolated complex-loaded AuNP. Finally, this vehicle obtained luciferase gene silencing in an animal tumor model.
Conclusion
Several multimolecular delivery vehicles are under clinical trial for RNAi-based cancer therapy but the dose amounts of siRNA (0.1-1.5mg/kg) are comparatively higher than levels observed in diseases of other organs (e.g. 0.15-0.3mg/kg in the liver). This may indicate that the highest expected RNAi efficacy in tumor is similar with that in the liver. RNAi efficacy in rapid-growing cancer cells is not comparable to relatively slow-growing hepatocytes because siRNA concentration in cytoplasm will dilute in divided cells. But more efficient delivery vehicles for tumor may contribute to increase RNAi rather than the present efficacies. Compared to a clinically approved Trastuzumab emtansine (half-life 1-4 days) and clinically tested anticancer drug loaded polymeric micelles (halflife 16-80h), the current clinically tested vehicles showed shorter circulation properties (half-life < 2h) (Table 3). This indicated that the current delivery vehicle need better performance. The clinical trial results and new biological evidences provide the clues for development of the next vehicle design.
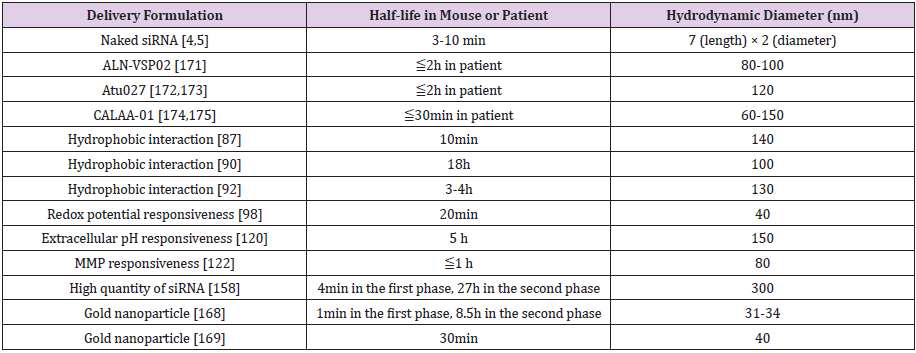
Table 3:Summary of blood circulation and size of delivery vehicles in this article [170].
a. The vehicle should exhibit long blood circulation
properties (half-life ≥2h). The higher amounts of circulating
delivery vehicle (containing siRNA) will increase possibility that the
vehicle diffuses/accumulates into tumor microenvironment. Some
vehicles introduced in this review showed longer than half-life =2h
but their doses for tumor growth inhibition in animal model were
not significantly lower than other vehicles [170-175]. These results
indicate that other aspects in vehicle design should be considered.
b. The vehicle should be smaller than 30nm diameter
size to enhance diffusion/ accumulation in tumor because the
nanoparticle with this diameter size penetrated in thick fibrotic
stroma and hypo vascular tumor in animal models. This research
indicates that less number of examples of Doxil (diameter size
90nm) in clinical tumor accumulation may be hampered by this size
limitation. Eventually, the behavior of delivery vehicle inside tumor
is governed by diffusion, implicating that smaller particles with less
than 30nm size are also preferred to reach cancer cells. Fabrication
of these small nanoparticles has gradually been realized by various
materials and techniques [176-179], e.g., unimer polyion complex/
gold nanoparticles and polymers [169,180-182]. Repeatedly,
we emphasize that size distribution of vehicle in buffer or fetal
bovine serum does not guarantee the same size distribution in
bloodstream.
c. Other functionalities (e.g. selective release of siRNA, high
cell specific recognition, and high endosome escapability)must
endow the delivery vehicle which simultaenously satisfied with
both (i) and (ii). To date, it is not clear which functionality is the
most critical factor to enhance RNAi in patient. Furthermore, the
delivery vehicle satisfied with (i) and (ii) but not (iii) does not
expect to exhibit superior RNAi than the current vehicles in clinical
trials. Ultimately, simpler formulation of delivery vehicles can be
more easily translated to their clinical use because of better quality
control as well as lower possibility of unexpected adverse effects.
References
- R Kanasty, JR Dorkin, A Vegas, D Anderson (2013) Delivery materials for siRNA therapeutics. Nat Mater 12(11): 967-977.
- HS Choi, W Liu, P Misra, E Tanaka, JP Zimmer, et al. (2007) Renal clearance of quantum dots. Nat Biotechnol 25(10): 1165-1170.
- A Schroeder, CG Levins, C Cortez, R Langer, DG Anderson (2010) Lipid-based nanotherapeutics for siRNA delivery. J Intern Med 267(1): 9-21.
- S Gao, F Dagnaes Hansen, EJB Nielsen, J Wengel, F Besenbacher, et al. (2009) The effect of chemical modification and nanoparticle formulation on stability and biodistribution of siRNA in mice. Mol Ther 17(7): 1225-1233.
- JJ Turner, SW Jones, SA Moschos, MA Lindsay, MJ Gait (2007) MALDI-TOF mass spectral analysis of siRNA degradation in serum confirms an RNAse A-like activity. Mol BioSyst 3: 43-50.
- Y Matsumura, H Maeda (1986) A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res 46(12): 6387-6392.
- H Cabral, J Makino, Y Matsumoto, P Mi, H Wu, et al. (2015) Systemic targeting of lymph node metastasis through the blood vascular system by using size-controlled nanocarriers. ACS Nano 9(5): 4957-4967.
- EA Sykes, J Chen, G Zheng, WCW Chan (2014) Investigating the Impact of Nanoparticle Size on Active and Passive Tumor Targeting Efficiency. ACS Nano 8(6): 5696-5706.
- X Zhu, Y Xu, LM Solis, W Tao, L Wang, et al. (2015) Long-circulating siRNA nanoparticles for validating prohibitin1-targeted non-small cell lung cancer treatment. Proc Natl Acad Sci USA 112(25): 7779-7784.
- MR Kano (2014) Nanotechnology and tumor microcirculation. Adv Drug Deliv Rev 74: 2-11.
- AE Hansen, AL Petersen, JR Henriksen, B Berresen, P Rasmussen, et al. (2015) Positron emission tomography based elucidation of the enhanced permeability and retention effect in dogs with cancer using copper-64 liposomes. ACS Nano 9: 6985-6995.
- F Yuan, M Dellian, D Fukumura, M Leunig, DA Berk, et al. (1995) Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res 55: 3752-3756.
- H Cabral, M Murakami, H Hojo, Y Terada, MR Kano, et al. (2013) Targeted therapy of spontaneous murine pancreatic tumors by polymeric micelles prolongs survival and prevents peritoneal metastasis. Proc Natl Acad Sci USA 110(28): 11397-11402.
- H Maeda (2015) Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv Drug Deliv Rev 91: 3-6.
- Ó Arrieta, LA Medina, E Estrada Lobato, N Hernández Pedro, G Villanueva Rodríguez, et al. (2012) First-line chemotherapy with liposomal doxorubicin plus cisplatin for patients with advanced malignant pleural mesothelioma: phase II trial. Br J Cancer 106(6): 1027-1032.
- JW Nicholas, YH Bae (2014) EPR: evidence and fallacy, J. Control. Release 190: 451-464.
- NM Marina, D Cochrane, E Harney, K Zomorodi, S Blaney, et al. (2002) Dose escalation and pharmacokinetics of pegylated liposomal doxorubicin (doxil) in children with solid tumos: a pediatric oncology group study. Clin Cancer Res 8(2): 413-418.
- A Hubert, O Lyass, D Pode, A Gabizon (2000) Doxil (Caelyx): an exploratory study with pharmacokinetics in patients with hormone-refractory prostate cancer, Anti-Cancer Drugs 11(2): 123-127.
- O Lyass, B Uziely, R Ben Yosef, D Tzemach, NI Heshing, et al. (2000) Correlation of toxicity with pharmacokinetics of pegylated liposomal doxorubicin (Doxil) in metastatic breast carcinoma. Cancer 89(5): 1037-1047.
- MJ Boers Sonderen, CM van Herpen, WT van der Graaf, IM Desar, MG van der Logt, et al. (2014) Correlation of toxicity and efficacy with pharmacokinetics (PK) of pegylated liposomal doxorubicin (PLD) (Caelyx®). Cancer Chemother Pharmacol 74: 457-463.
- Z Symon, A Peyser, D Tzemach, O Lyass, E Sucher, et al. (1999) Selective delivery of doxorubicin to patients with breast carcinoma metastases by stealth liposomes. Cancer 86(1): 72-78.
- T Lammers, F Kiessling, WE Hennink, G Storm (2012) Drug targeting to tumors: principles, pitfalls, and (pre-) clinical progress. J Control Release 161(2): 175-187.
- YH Bae, K Park (2011) Targeted drug delivery to tumors: myths, reality, and possibility. J Control Release 153(3): 198-205.
- S Taurin, H Nehoff, K Greish (2012) Anticancer nanomedicine and tumor vascular permeability; where is the missing link? J Control Release 164(3): 265-275.
- Y Matsumoto, JW Nichols, K Toh, T Nomoto, H Cabral, et al. (2016) Vascular bursts enhance permeability of tumor blood vessels and improve nanoparticle delivery. Nat Nanotechol 11(6): 533-538.
- VP Chauhan, RK Jain (2013) Strategies for advancing cancer nanomedicine. Nat Mater 12(11): 958-962.
- NR Smith, D Baker, M Farren, A Pommier, R Swann, et al. (2013) Tumor stromal architecture can define the intrinsic tumor response to VEGF-targeted therapy. Clin Cancer Res 19(24): 6943-6956.
- SD Perrault, C Walkey, T Jennings, HC Fischer, WC Chan (2009) Mediating tumor targeting efficiency of nanoparticles through design. Nano Lett 9(5): 1909-1915.
- Z Popović, W Liu, VP Chauhan, J Lee, C Wong, et al. (2010) A nanoparticle size series for in vivo fluorescence imaging. Angew Chem Int Ed 49(46): 8649-8652.
- RK Jain, JD Martin, T Stylianopoulos (2014) The role of mechanical forces in tumor growth and therapy. Annu Rev Biomed Eng 16: 321-346.
- MR Kano, Y Bae, C Iwata, Y Morishita, M Yashiro, et al. (2007) Improvement of cancer-targeting therapy, using nanocarriers for intractable solid tumors by inhibition of TGF-β Proc Natl Acad Sci U S A 104(9): 3460-3465.
- TD Mckee, P Grandi, W Mok, G Alexandrakis, N Insin, et al. (2006) Degradation of fibrillary collagen in a human melanoma xenograft improves the efficacy of an oncolytic herpes simplex virus vector. Cancer Res 66(5): 2509-2513.
- YS Kanwar, MG Farquhar (1979) Presence of heparan sulfate in the glomerular basement membrane. Proc Natl Acad Sci USA 76(3): 1303-1307.
- T Soji, Y Murata, A Ohira, H Nishizono, M Tanaka, et al. (1992) Evidence that hepatocytes can phagocytize exogenous substances. Anat Rec 233(4): 543-546.
- K Elvevold, B Smedsrød, I Martinez (2008) The liver sinusoidal endothelial cell: a cell type of controversial and confusing identity. Am J Physiol Gastrointest Liver Physiol 294(2): G391-G400.
- HH Gustafson, D Holt Casper, DW Grainger, H Ghandehari (2015) Nanoparticle uptake: the phagocyte problem. Nano Today 10(4): 487-510.
- K Braeckmans, K Buyens, W Bouquet, C Vervaet, P Joye, et al. (2010) Sizing nano matter in biological fluids by fluorescence single particle tracking. Nano Lett 10(11): 4435-4442.
- A Akinc, G Battaglia (2013) Exploiting endocytosis for nanomedicines. Cold Spring Harb Perspect Biol 5(11): a016980.
- EM Pelegri O'Day, EW Lin, HD Maynard (2014) Therapeutic protein-polymer conjugates: advancing beyond PEGylation. J Am Chem Soc 136(41): 14323-14332.
- K Knop, R Hoogenboom, D Fischer, US Schubert (2010) Poly(ethylene glycol) in drug delivery: pros and cons as well as potential alternatives. Angew Chem Int Ed 49(36): 6288-6308.
- Y Qi, A Chilkoti (2015) Protein-polymer conjugation-moving beyond PEgylation. Curr Opin Chem Biol 28: 181-193.
- Y Barenholz (2012) Doxil®-the first FDA-approved nano-drug: lessons learned. J Control Release 160(2): 117-134.
- E Pérez Herrero, A Fernández Medarde (2015) Advanced targeted therapies in cancer: drug nanocarriers, the future of chemotherapy. Eur J Pharm Biopharm 93: 52-79.
- H Schellekens, WE Hennink, V Brinks (2013) The immunogenicity of polyethylene glycol: facts and fiction. Pharm Res 30(7): 1729-1734.
- N Longo, CO Harding, BK Burton, DK Grange, J Vockley, et al. (2014) Single-dose, subcutaneous recombinant phenylalanine ammonia lyase conjugated with polyethylene glycol in adult patients with phenylketonuria: an open-label, multicenter, phase 1 dose-escalation trial. Lancet 384(9937): 37-44.
- Q Yang, SK Lai (2015) Anti-PEG immunity: emergence, characteristics, and unaddressed questions. Wiley Interdiscip Rev Nanomed Nanobiotechnol 7(5): 655-677.
- JK Armstrong, G Hempel, S Koling, LS Chan, T Fisher, et al. (2007) Antibody against poly(ethylene glycol) adversely affects PEG-asparaginase therapy in acute lymphoblastic leukemia patients. Cancer 110(1): 103-111.
- CM Beddoes, CP Case, WH Briscoe (2015) Understanding nanoparticle cellular entry: A physicochemical perspective. Adv Colloid Interf Sci 218: 48-68.
- I Canton, G Battaglia (2012) Endocytosis at the nanoscale. Chem Soc Rev 41: 2718-2739.
- S Venkataraman, JL Hedrick, ZY Ong, C Yang, PL Ee, et al. (2011) The effects of polymeric nanostructure shape on drug delivery. Adv Drug Deliv Rev 63(14-15): 1228-1246.
- JD Trono, K Mizuno, N Yusa, T Matsukawa, K Yokoyama, et al. (2011) Size, concentration and incubation time dependence of gold nanoparticle uptake into pancreas cancer cells and its future application to X-ray drug delivery system. J Radiat Res 52(1): 103-109.
- DB Chithrani (2010) Intracellular uptake, transport, and processing of gold nanostructures. Mol Membr Biol 27(7): 299-311.
- H Gao, W Shi, LB Freund (2005) Mechanics of receptor-mediated endocytosis. Proc Natl Acad Sci USA 102(27): 9469-9474.
- ZU Rehman, IS Zuhorn, D Hoekstra (2013) How cationic lipids transfer nucleic acids into cells and across cellular membranes: recent advances. J Control Release 166(1): 46-56.
- K Itaka, K Kataoka (2009) Recent development of nonviral gene delivery systems with virus-like structures and mechanism. Eur J Pharm Biopharm 71(3): 475-483.
- N Bertrand, J Wu, X Xu, N Kamaly, OC Farokhza (2014) Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev 66: 2-25.
- Y Zhong, F Meng, C Deng, Z Zhong (2014) Ligand-directed active tumor-targeting polymeric nanoparticles for cancer chemotherapy. Biomacromolecules 15(6): 1955-1969.
- K Temming, RM Schiffelers, G Molema, RJ Kok (2005) RGD-based strategies for selective delivery of therapeutics and imaging agents to the tumor vasculature. Drug Resist Updat 8(6): 381-402.
- M Sutherland, A Gordon, SD Shnyder, LH Patterson, HM Sheldrake (2012) RGD binding integrins in prostate cancer: expression patterns and therapeutic prospects against bone metastasis. Cancers 4(4): 1106-1145.
- L Sancey, E Garanger, S Foilard, G Schoehn, A Hurbin, et al. (2009) Clustering and internalization of integrin αvβ3 with a tetrameric RGD-synthetic peptide. Mol Ther 17(5): 837-843.
- G Trapani, N Denora, A Trapani, V Laquintana (2012) Recent advances in ligand targeted therapy. J Drug Target 20(1): 1-22.
- KE Tiller, PM Tessier (2015) Advances in antibody design. Annu Rev Biomed Eng 17: 191-216.
- C Peters, S Brown (2015) Antibody-drug conjugates as novel anti-cancer chemotherapeutics. Biosci Rep 35(4): e00225.
- N Zhang, L Liu, CD Dumitru, NR Cummings, M Cukan, et al. (2011) Glycoengineered pichia produced anti-HER2 is comparable to trastuzumab in preclinical study. mAbs 3(3): 289-298.
- L Elmlund, C Kӓck, T Aastrup, IA Nicholls (2015) Study of the interaction of trastuzumab and SKOV3 eptithelial cancer cells using a quartz crystal microbalance sensor. Sensors 15(3): 5884-5894.
- H Tong, L Zhang, A Kaspar, MJ Rames, L Huang, et al. (2013) Peptide conjugation induced conformational changes in human IgG1 observed by optimized negative-staining and individual-particle electron tomography. Sci Rep 3: 1089.
- R van der Meel, LJ Vehmeijer, RJ Kok, G Storm, EV van Gaal (2013) Ligand-targeted particulate nanomedicines undergoing clinical evaluation: current status. Adv Drug Deliv Rev 65(10): 1284-1298.
- JA Reddy, R Dorton, E Westrick, A Dawson, T Smith, et al. (2007) Preclinical evaluation of EC145, a folate-Vinca alkaloid conjugate. Cancer Res 67(9): 4434-4442.
- RW Naumann, RL Coleman, RA Burger, EA Sausville, E Kutarska, et al. (2013) Precedent: a randomized phase II trial comparing vintafolide (EC145) and pegylated liposomal doxorubicin (PLD) in combination versus PLD alone in patient with platinum-resistant ovarian cell. J Clin Oncol 31(35): 4400-4406.
- K Park (2013) Facing the truth about nanotechnology in drug delivery. ACS Nano 7(9): 7442-7447.
- JA Mindell (2012) Lysosomal acidification mechanisms. Annu Rev Physiol 74: 69-86.
- H Appelqvist, P Wäster, K Kågedal, K Öllinger (2013) The lysosome: from waste bag to potential therapeutic target. J Mol Cell Biol 5(4): 214-226.
- S Höbel, A Aigner (2013) Polyethylenimines for siRNA andmiRNA delivery in vivo. Wiley Interdiscip Rev Nanomed Nanobiotechnol 5(5): 484-501.
- K Miyata, M Oba, M Nakanishi, S Fukushima, Y Yamasaki, et al. (2008) Polyplexes from poly(aspartamide) bearing 1,2-diaminoethane side chains induce pH-selective endosomal membrane destabilization with amplified transfection and negligible cytotoxicity. J Am Chem Soc 130(48): 16287-16294.
- JP Behr (1997) The proton sponge: a trick to enter cells the viruses did not exploit. Chimia 51(1-2): 34-36.
- ND Sonawane, FC Szoka, AS Verkman (2003) Chloride accumulation and swelling in endosomes enhances DNA transfer by polyamine-DNA polyplexes. J Biol Chem 278(45): 44826-44831.
- K Miyata, N Nishiyama, K Kataoka (2012) Rational design of smart supramolecular assemblies for gene delivery: chemical challenges in the creation of artificial viruses. Chem Soc Rev 41: 2562-2574.
- NM Rao (2010) Cationic lipid-mediated nucleic acid delivery: beyond being cationic. Chem Phys Lipids 163(3): 245-252.
- K Kataoka, A Harada, Y Nagasaki (2001) Block copolymer micelles for drug delivery: design, characterization and biological significance. Adv Drug Deliv Rev 47(1): 113-131.
- Y Dong, KT Love, JR Dorkin, S Sirirungruang, Y Zhang, et al. (2014) Lipopeptide nanoparticles for potent and selective siRNA delivery in rodents and nonhuman primates. Proc Natl Acad Sci USA 111(11): 3955-3960.
- D Schaffert, C Troiber, EE Salcher, T Fröhlich, I Martin, et al. (2011) Solid-phase synthesis of sequence-defined T-, i-, and U-shape polymers for pDNA and siRNA delivery. Angew Chem Int Ed 50(38): 8986-8989.
- TK Endres, M Beck Broichsitter, O Samsonova, T Renette, TH Kissel (2011) Self-assembled biodegradable amphiphilic PEG-PCL-lPEI triblock copolymers at the borderline between micelles and nanoparticles designed for drug and gene delivery. Biomaterials 32(30): 7721-7731.
- TM Sun, JZ Du, YD Yao, CQ Mao, S Dou, et al. (2011) Simultaneous delivery of siRNA and paclitaxel via a “two-in-one” micelleplex promotes synergistic tumor suppression. ACS Nano 5(2): 1483-1494.
- H Yu, Y Zou, Y Wang, X Huang, G Huang, et al. (2011) Overcoming endosomal barrier by amphotericin B-loaded dual pH-responsive PDMA-b-PDPA micelleplexes for siRNA delivery. ACS Nano 5(11): 9246-9255.
- HJ Kim, A Ishii, K Miyata, Y Lee, S Wu, et al. (2010) Introduction of stearoyl moieties into a biocompatible cationic polyaspartamide derivative, PAsp(DET), with endosomal escaping function for enhanced siRNA-mediated gene knockdown. J Control Release 145(2): 141-148.
- A Philipp, X Zhao, P Tarcha, E Wagner, A Zintchenko (2009) Hydrophobically modified oligoethylenimines as highly efficient transfection agents for siRNA delivery. Bioconjug Chem 20(11): 2055-2061.
- HJ Kim, M Oba, F Pittella, T Nomoto, H Cabral, et al. (2012) PEG-detachable cationic polyaspartamide derivatives bearing stearoyl moieties for systemic siRNA delivery toward subcutaneous BxPC3 pancreatic tumor. J Drug Target 20(1): 33-42.
- HJ Kim, T Ishii, M Zheng, S Watanabe, K Toh, et al. (2014) Multifunctional polyion complex micelle featuring enhanced stability, targetability, and endosome escapability for systemic siRNA delivery to subcutaneous model of lung cancer. Drug Deliv Transl Res 4(1): 50-60.
- HJ Kim, K Miyata, T Nomoto, M Zheng, A Kim, et al. (2014) siRNA delivery from triblock copolymer micelles with spatially-ordered compartments of PEG shell, siRNA-loaded intermediate layer, and hydrophobic core. Biomaterials 35(15): 4548-4556.
- N Yagi, I Manabe, T Tottori, A Ishihara, F Ogata, et al. (2009) A nanoparticle system specifically designed to deliver short interfering RNA inhibits tumor growth in vivo. Cancer Res 69(16): 6531-6538.
- BL Mui, YK Tam, M Jayaraman, SM Ansell, X Du, et al. (2013) Influence of polyethylene glycol lipid desorption rates on pharmacokinetics and pharmacodynamics of siRNA lipid nanoparticles. Mol Ther Nucleic Acids 2: e139.
- Q Sun, Z Kang, L Xue, Y Shang, Z Su, et al. (2015) A collaborative assembly strategy for tumor-targeted siRNA delivery. J Am Chem Soc 137(18): 6000-6010.
- JM Hansen, C Harris (2014) Glutathione during embryonic development. Biochim Biophys Acta 1850(8): 1527-1542.
- ML Circu, TY Aw (2012) Glutathione and modulation of cell apoptosis. Biochim Biophys Acta 1823(10): 1767-1777.
- M Smeyne, RJ Smeyne (2013) Glutathione metabolism and Parkinson's disease. Free Radic Biol Med 62: 13-25.
- JH Ryu, RT Chacko, S Jiwpanich, S Bickerton, RP Babu, et al. (2010) Self-cross-linked polymer nanogels: a versatile nanoscopic drug delivery platform. J Am Chem Soc 132(48): 17227-17235.
- S Matsumoto, RJ Christie, N Nishiyama, K Miyata, A Ishii, et al. (2009) Environment-responsive block copolymer micelles with a disulfide cross-linked core for enhanced siRNA delivery. Biomacromolecules 10(1): 119-127.
- Y Oe, RJ Christie, M Naito, SA Low, S Fukushima, et al. (2014) Actively-targeted polyion complex micelles stabilized by cholesterol and disulfide cross-linking for systemic delivery of siRNA to solid tumors. Biomaterials 35(27): 7887-7895.
- DB Rozema, DL Lewis, DH Wakefield, SC Wong, JJ Klein, et al. (2007) Dynamic polyconjugates for targeted in vivo delivery of siRNA to hepatocytes. Proc Natl Acad Sci USA 104(32): 12982-12987.
- RG Gish, MF Yuen, HLY Chan, BD Given, CL Lai, et al. (2015) Synthetic RNAi triggers and their use in chronic hepatitis B therapies with curative intent. Antivir Res 121: 97-108.
- T Stauber, TJ Jentsch (2013) Chloride in vesicular trafficking and function. Annu Rev Physiol 75: 453-477.
- N Murthy, YX Thng, S Schuck, MC Xu, JMJ Fréchet (2002) A novel strategy for encapsulation and release of proteins: hydrogels and microgels with acid-labile acetal cross-linkers. J Am Chem Soc 124(42): 12398-12399.
- Y Bae, S Fukushima, A Harada, K Kataoka (2003) Design of environment-sensitive supramolecular assemblies for intracellular drug delivery: polymeric micelles that are responsive to intracellular pH change. Angew Chem Int Ed 42(38): 4640-4643.
- M Oishi, Y Nagasaki, K Itaka, N Nishiyama, K Kataoka (2005) Lactosylated poly(ethylene glycol)-siRNA conjugate through acid-labile β-thiopropionate linkage to construct pH-sensitive polyion complex micelles achieving enhanced gene silencing in hepatoma cells. J Am Chem Soc 127(6): 1624-1625.
- JH Jeong, SW Kim, TG Park (2003) Novel intracellular delivery system of antisense oligonucleotide by self-assembled hybrid micelles composed of DNA/PEG conjugate and cationic fusogenic peptide. Bioconjug Chem 14(2): 473-479.
- J Heller, J Barr, SY Ng, KS Abdellauoi, R Gurny (2002) Poly(ortho esters): synthesis, characterization, properties and uses. Adv Drug Deliv Rev 54(7): 1015-1039.
- Y Lee, S Fukushima, Y Bae, S Hiki, T Ishii, et al. (2007) A protein nanocarrier from charge-conversion polymer in response to endosomal pH. J Am Chem Soc 129(17): 5362-5363.
- Y Lee, T Ishii, H Cabral, HJ Kim, JH Seo, et al. (2009) Charge-conversional polyionic complex micelles-efficient nanocarriers for protein delivery into cytoplasm. Angew Chem Int Ed 48(29): 5309-5312.
- MS Shim, YJ Kwon (2009) Acid-responsive linear polyethyleneimine for efficient, specific, and biocompatible siRNA delivery. Bioconjug Chem 20(3): 488-499.
- H Takemoto, K Miyata, S Hattori, T Ishii, T Suma, et al. (2013) Acidic pH-responsive siRNA conjugate for reversible carrier stability and accelerated endosomal escape with reduced IFNα-associated immune response. Angew Chem Int Ed 52(24): 6218-6221.
- A Kulkarni, K DeFrees, SH Hyun, DH Tompson (2012) Pendant polyer:amino-β-cyclodextrin: siRNA guest:host nanoparticles as efficient vectors for gene silencing. J Am Chem Soc 134(18): 7596-7599.
- Y Nie, M Günther, Z Gu, E Wagner (2011) Pyridylhydrazone-based PEGylation for pH-reversible lipopolyplex shielding. Biomaterials 32(3): 858-869.
- XZ Yang, JZ Du, S Dou, CQ Mao, HY Long, et al. (2012) Sheddable ternary nanoparticles for tumor acidity-targeted siRNA delivery. ACS Nano 6(1): 771-781.
- MW Gorman, EO Feigl, CW Buffington (2007) Human plasma ATP concentration. Clin Chem 53(2): 318-325.
- M Naito, T Ishii, A Matsumoto, K Miyata, Y Miyahara, et al. (2012) A phenylboronate-functionalized polyion complex micelle for ATP-triggered release of siRNA. Angew Chem Int Ed 51(43): 1-6.
- R Mo, T Jiang, R DiSanto, W Tai, Z Gu (2014) ATP-triggered anticancer drug delivery. Nat Commun 5: 3364.
- R Mo, T Jiang, Z Gu (2014) Enhanced anticancer efficacy by ATP-mediated liposomal drug delivery. Angew Chem 126(23): 5925-5930.
- DF Quail, JA Joyce (2013) Microenvironmental regulation of tumor progression and metastasis. Nat Med 19(11): 1423-1437.
- D Neri, CT Supuran (2011) Interfering with pH regulation in tumors as a therapeutic strategy. Nat Rev Drug Discov 10(10): 767-777.
- J Li, X Yu, Y Wang, Y Yuan, H Xiao, et al. (2014) A reduction and pH dual sensitive polymeric vector for long-circulating and tumor-targeted siRNA delivery. Adv Mater 26(48): 8217-8224.
- S Hama, S Itakura, M Nakai, K Nakayama, S Morimoto, et al. (2015) Overcoming the polyethylene glycol dilemma via pathological environment-sensitive change of the surface property of nanoparticles for cellular entry. J Control Release 206: 67-74.
- HX Wang, XZ Yang, CY Sun, CQ Mao, YH Zhu, et al. (2014) Matrix metalloproteinase 2-responsive micelle for siRNA delivery. Biomaterials 35(26): 7622-7634.
- LA Shuman Moss, S Jensen Taubman, WG Stetler Stevenson (2012) Matrix metalloproteinases: changing roles in tumor progression and metastasis. Am J Pathol 181(6): 1895-1899.
- J Decock, S Thirkettle, L Wagstaff, DR Edwards (2011) Matrix metalloproteinases: protective roles in cancer. J Cell Mol Med 15(6): 1254-1265.
- K Kessenbrock, V Plaks, Z Werb (2010) Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 141(1): 52-67.
- H Li, SS Yu, M Miteva, CE Nelson, T Werfel, et al. (2013) Matrix metalloproteinase responsive, proximity-activated polymeric nanoparticles for siRNA delivery. Adv Funct Mater 23(24): 3040-3052.
- H Li, M Miteva, KC Kirkbride, MJ Cheng, CE Nelson, et al. (2015) Dual MMP7-proximity-activated and folate receptor-targeted nanoparticles for siRNA delivery. Biomacromolecules 16(1): 192-201.
- H Hatakeyama, H Akita, E Ito, Y Hayashi, M Oishi, et al. (2011) Systemic delivery of siRNA to tumors using a lipid nanoparticle containing a tumor-specific cleavable PEG-lipid. Biomaterials 32(18): 4306-4316.
- L Zhu, T Wang, F Perche, A Taigind, VP Torchilin (2013) Enhanced anticancer acitivty of nano preparation containing an MMP2-sensitive PEG-drug conjugate and cell penetrating moiety. Proc Natl Acad Sci USA 110(42): 17047-17052.
- S Huang, K Shao, Y Kuang, Y Liu, J Li, et al. (2013) Tumor targeting and microenvironment-responsive nanoparticles for gene delivery. Biomaterials 34(21): 5294-5302.
- SH van Rijt, DA Bölükbas, C Argyo, S Datz, M Lindner, et al. (2015) Protease-mediated release of chemotherapeutics from mesoporous silica nanoparticles to ex vivo human and mouse lung tumors. ACS Nano 9(3): 2377-2389.
- J Le, Z Ge, S (2013) Liu PEG-sheddable polyplex micelles as smart gene carriers based on MMP-cleavable peptide-linked block copolymers. Chem Commun 49: 6974-6976.
- W Gao, B Xiang, TT Meng, F Liu, XR Qi (2013) Chemotherapeutic drug delivery to cancer cells using a combination of folate targeting and tumor microenvironment-sensitive polypeptides. Biomaterials 34(16): 4137-4149.
- JK Nair, JLS Willoughby, A Chan, K Charisse, MR Alam, et al. (2014) Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J Am Chem Soc 136(49): 16958-16961.
- BJ Zern, AM Chacko, J Liu, CF Greineder, ER Blankemeyer, et al. (2013) Reduction of nanoparticle avidity enhances the selectivity of vascular targeting and PEG detection of pulmonary inflammation. ACS Nano 7(3): 2461-2469.
- LJ Cruz, PJ Tacken, R Fokkink, CG Figdor (2011) The influence of PEG chain length and targeting moiety on antibody-mediated delivery of nanoparticle vaccines to human dendritic cells. Biomaterials 32(28): 6791-6803.
- JF Stefanick, JD Ashley, T Kiziltepe, B Bilgicer (2013) A systemic analysis of peptide linker length and liposomal polyethylene glycol coating on cellular uptake of peptide targeted liposomes. ACS Nano 7(4): 2935-2947.
- S Hak, E Helgesen, HH Hektoen, EM Huuse, PA Jarzyna, et al. (2012) The effect of nanoparticle polyethylene glycol surface density on ligand-directed tumor targeting studied in vivo by dual modality imaging. ACS Nano 6(6): 5648-5658.
- EA Sykes, J Chen, G Zheng, W Chan (2014) Investigating the impact of nanoparticle size on active and passive tumor targeting efficiency. ACS Nano 8(6): 5696-5706.
- C Wolfrum, S Shi, KN Jayaprakash, M Jayaraman, G Wang, et al. (2007) Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nat Biotechnol 25(10): 1149-1157.
- J Soutschek, A Akinc, B Bramlage, K Charisse, R Constien, et al. (2004) Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 432(7041): 173-178.
- OF Khan, EW Zaia, H Yin, RL Bogorad, JM Pelet, et al. (2014) Ionizable amphiphilic dendrimer-based nanomaterials with alkyl-chain-substituted amines for tunable siRNA delivery to the liver endothelium in vivo. Angew Chem Int Ed 53(52): 1-6.
- RG Smits, GJM Koper, M Mandel (1993) The influence of nearest- and next-nearest neighbor interactions on the potentiometric titration of linear poly(ethylenimine). J Phys Chem 97(21): 5745-5751.
- Y Lee, H Mo, H Koo, JY Park, MY Cho, et al. (2007) Visualization of the degradation of a disulfide polymer, linear poly(ethylenimine sulfide), for gene delivery. Bioconjug Chem 18(1): 13-18.
- MA Gosselin, W Guo, RJ Lee (2001) Efficient gene transfer using reversible cross-linked low molecular weight polyethylenimine. Bioconjug Chem 12(6): 989-994.
- H Uchida, K Miyata, M Oba, T Ishii, T Suma, et al. (2011) Odd-even effect of repeating aminoethylene units in the side chain of N-substituted polyaspartamides on gene transfection profiles. J Am Chem Soc 133(39): 15524-15532.
- F Pittella, K Miyata, Y Maeda, T Suma, S Watanabe, et al. (2012) Pancreatic cancer therapy by systemic administration of VEGF siRNA contained in calcium phosphate/charge-conversional polymer hybrid nanoparticles. J Control Release 161(3): 868-874.
- AJ Convertine, DSW Benoit, CL Duvall, AS Hoffman, PS Stayton (2009) Development of a novel endosomolytic diblock copolymer for siRNA delivery. J Control Release 133(3): 221-229.
- P Midoux, C Pichon, JJ Yaouanc, PA Jaffrés (2009) Chemical vectors for gene delivery: a current review on polymers, peptides and lipids containing histidine or imidazole as nucleic acids carriers. Br J Pharmacol 157(2): 166-178.
- JE Dahlman, C Barnes, OF Khan, A Thiriot, S Jhunjunwala, et al. (2014) In vivo endothelial siRNA delivery using polymeric nanoparticles with low molecular weight. Nat Biotechnol 9(8): 648-655.
- F Perche, S Biswas, T Wang, L Zhu, VP Torchilin (2014) Hypoxia-targeted siRNA delivery. Angew Chem Int Ed 53(13): 3362-3366.
- Y Xu, FC Szoka (1996) Mechanism of DNA release from cationic liposome/DNA complexes used in cell transfection. Biochemistry 35(18): 5616-5623.
- H Hatakeyama, H Akita, H Harashima (2011) A multifunctional envelope type nano device (MEND) for gene delivery to tumours based on the EPR effect: a strategy for overcoming the PEG dilemma. Adv Drug Deliv Rev 63(3): 152-160.
- IM Hafez, N Maurer, PR Cullis (2001) On the mechanism whereby cationic lipids promote intracellular delivery of polynucleic acids. Gene Ther 8(15): 1188-1196.
- CE Ashley, EC Carnes, KE Epler, DP Padilla, GK Phillips, et al. (2012) Delivery of small interfering RNA by peptide-targeted mesoporous silica nanoparticle supported lipid bilayers. ACS Nano 6(3): 2174-2188.
- A Elbakry, A Zaky, R Liebl, R Rachel, A Geopferich, et al. (2009) Layer-by-layer assembled gold nanoparticles for siRNA delivery. Nano Lett 9(5): 2059-2064.
- SK Lee, CH Tung (2013) A Fabricated siRNA nanoparticle for ultralong gene silencing in vivo. Adv Funct Mater 23(28): 3488-3493.
- ZJ Deng, SW Morton, E Ben Akiva, EC Dreaden, KE Shopsowitz, et al. (2013) Layer-by-layer nanoparticles for systemic codelivery of an anticancer drug and siRNA for potential triple-negative breast cancer treatment. ACS Nano 7(11): 9571-9584.
- HM Coley (2008) Mechanisms and strategies to overcome chemotherapy resistance in metastatic breast cancer. Cancer Treat Rev 34(4): 378-390.
- M Taheri, F Mahjoubi (2013) MRP1 but not MDR1 is associated with response to neoadjuvant chemotherapy in breast cancer patients. Dis Markers 34(6): 387-393.
- NS Gandhi, RK Tekade, MB Chougule (2014) Nanocarrier mediated delivery of siRNA/miRNA in combination with chemotherapeutic agents for cancer therapy: current progress and advances. J Control Release 194: 238-256.
- T Suma, K Miyata, Y Anraku, S Watanabe, RJ Christie, et al. (2012) Smart multilayered assembly for biocompatible siRNA delivery featuring dissolvable silica, endosome-disrupting polycations, and detachable PEG. ACS Nano 6(8): 6693-6705.
- Y Kakizawa, S Furukawa, A Ishii, K Kataoka (2006) Organic-inorganic hybrid nanocarrier of siRNA constructing through the self-assembly of calcium phosphate and PEG-based block aniomer. J Control Release 111(3): 368-370.
- F Pittella, M Zhang, Y Lee, HJ Kim, T Tockary, et al. (2011) Enhanced endosomal escape of siRNA-incorporating hybrid nanoparticles from calcium phosphate and PEG-block charge-conversional polymer for efficient gene knockdown with negligible cytotoxicity. Biomaterials 32(11): 3106-3114.
- M Yamaguchi (2014) Regulatory role of regucalcin in heart calcium signaling: insight into cardiac failure (Review). Biomed Rep 2(3): 303-308.
- J Li, YC Chen, YC Tseng, S Mozumdar, L Huang (2010) Biodegradable calcium phosphate nanoparticle with lipid coating for systemic siRNA delivery. J Control Release 142(3): 416-421.
- Y Yang, J Li, F Liu, L Huang (2012) Systemic delivery of siRNA via LCP nanoparticle efficiently inhibits lung metastasis. Mol Ther 20(3): 609-615.
- SA Jensen, ES Day, CH Ko, LA Hurley, JP Luciano, et al. (2013) Spherical nucleic acid nanoparticle conjugates as an RNAi-based therapy for glioblastoma. Sci Transl Med 5(209): 209ra152.
- HJ Kim, H Takemoto, Y Yi, M Zheng, Y Maeda, et al. (2014) Precise engineering of siRNA delivery vehicles to tumors using polyion complexes and gold nanoparticles. ACS Nano 8(9): 8979-8991.
- Hyun Jin Kim, Ahram Kim, Kanjiro Miyata, Kazunori Kataoka (2016) Recent progress in development of siRNA delivery vehicles for cancer therapy. Advanced Drug Delivery Reviews 104: 61-77.
- J Tabernero, GI Shapiro, PM LoRusso, A Cervantes, GK Schwartz, et al. (2013) First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov 3(4): 406-417.
- B Schultheis, D Strumberg, AC Vank, F Gebhardt, O Keil, et al. (2014) First-in-human phase I study of the liposomal RNA interference therapeutic Atu027 in patients with advanced solid tumors. J Clin Oncol 32(36): 4141-4148.
- A Santel, M Aleku, O Keil, J Endruschat, V Esche, et al. (2006) A novel siRNA-lipoplex technology for RNA interference in the mouse vascular endothelium. Gene Ther 13: 1222-1234.
- DW Bartlett, ME Davis (2007) Physiochemical and biological characterization of targeted, nucleic acid-containing nanoparticles. Bioconjug Chem 18(2): 456-468.
- JE Zuckerman, I Gritli, A Tolcher, JD Heidel, D Lim, et al. (2014) Correlating animal and human phase Ia/Ib clinical data with CALAA-01, a targeted, polymer-based nanoparticle containing siRNA. Proc Natl Acad Sci USA 111(31): 11449-11454.
- Loutfy H Madkour (2020) Reactive Oxygen Species (ROS), Nanoparticles, and Endoplasmic Reticulum (ER) Stress-Induced Cell Death Mechanisms. In: Loutfy H Madkour (Edt.)., Imprint: Academic Press Published, USA. pp.780.
- Loutfy H Madkour (2020) Nanoparticles Induce Oxidative and Endoplasmic Reticulum Antioxidant Therapeutic Defenses. In: Loutfy H Madkour (Edt.)., Springer Nature Switzerland.
- Loutfy H Madkour (2020) Nucleic Acids as Gene Anticancer Drug Delivery Therapy. In: Loutfy H Madkour (Edt.)., (1st)., Imprint: Academic Press Paperback, USA. pp.650.
- Loutfy H Madkour (2019) Nanoelectronic Materials: Fundamentals and Applications (Advanced Structured Materials). In: Loutfy H Madkour (Edt.)., (1st)., Springer Nature Switzerland AG.
- H Shimizu, Y Hori, S Kaname, K Yamada, N Nishiyama, et al. (2010) siRNA-based therapy ameliorates glomerulonephritis. J Am Soc Nephrol 21(4): 622-633.
- A Sizovs, X Song, MN Waxham, Y Jia, F Feng, et al. (2014) Precisely tunable engineering of sub-30 nm monodisperse oligonucleotide nanoparticles. J Am Chem Soc 136(1): 234-240.
- C Dohmen, D Edinger, T Fröhlich, L Schreiner, U Lächelt, et al. (2012) Nanosized multifunctional polyplexes for receptor-mediated siRNA delivery. ACS Nano 6(6): 5198-5208.
