 Research Article
Research ArticleAbstract
Aging is a biological process during lifespan with accumulation of mutations and damages, lowering fitness of at older ages and increasing hazards to survival. Aging is the most important risk factor associated with many diseases, such as cardiovascular disease, cancer, type 2 diabetes, hypertension, and Alzheimer’s disease. It accounts for about two thirds of death world-wide, and an even higher rate of 90% in developed countries. Understanding the biological mechanism of aging will therefore lead to tremendous public health benefits. Epigenetic markers, which refers to changes to the genome that do not involve changes in DNA sequence, have gain popularity in recent research by building connections between genetic and environmental aging factors. These epigenetic markers, e.g., telomere length and DNA methylation, can lead to a pre-diction of biological age or acceleration of aging, which can be further related to age-related diseases. A long-term goal of this project is to build an effective prediction algorithm for biological age using epigenetic markers. As the first step, a literature review was conducted on public database for existing studies on telomere length and all-cause mortality to prove the concept. A total of 27 studies were found in the period of 2003-2019. A weighted z-score metaanalysis was performed to assess the association be-tween leukocytes telomere length and all-cause mortality (p-value = 6.8E-14). A preliminary analysis of DNA methylation markers was also run with all-cause mortality as an alternative epigenetic marker. Results from 15 studies were combined using random-effect meta-analysis (p-value < 1E-308).
Keywords: Aging; Meta-Analysis; Telomere Length; DNA Methylation
Abbreviations: SB: Southern Blotting; qPCR: Quantitative Polymerase Chain Reaction; Cis: Confidence Intervals; SE: Standard Error; HR: Hazard Ratio
Introduction
Aging is a biological process during lifespan with accumulation of mutations and damages, lowering fitness of at older ages and increasing hazards to survival [1]. Aging is the most important risk factor associated with many diseases, such as cardiovascular disease, cancer, type 2 diabetes, hypertension, and Alzheimer’s disease [2]. It accounts for about two thirds of death worldwide, and an even higher rate of 90% in developed countries. Understanding the biological mechanism of aging will therefore lead to tremendous public health benefits. Aging process can be affected by both genetic and nongenetic factors. The nongenetic intervention on aging can be long-lasting, and potentially explained by epigenetic mechanisms. Epigenetics, which refers to changes to the genome that do not involve changes in DNA sequence, have gain popularity in recent research [3]. These epigenetic markers, e.g., telomere length and DNA methylation, can lead to a prediction of biological age or acceleration of aging, and age-related diseases [4]. Telomeres are repetitive noncoding DNA components located at the end of chromosomes to protect from degradation of coding sequences.
The telomeres shorten each time a cell divides because of the end replication problem, but also by oxidative stress, and lengthened by the enzyme telomerase and DNA exchange during mitosis [5,6]. Telomere attrition has been widely reported to be associated with increased morbidity and mortality of various age-related diseases [7]. DNA methylation is a process by which methyl groups are added to the DNA molecule. Methylation can change the activity of a DNA segment without changing the sequence [8,9]. Recently developed indices of cellular age based on DNA methylation data are being used to study factors that influence the rate of aging and the health correlates of these metrics of the epigenetic clock [10]. This paper uses meta-analysis to predict the relationship between two epigenetic markers, telomere length and DNA methylation, and all-cause mortality [11]. The telomere length meta-analysis is based on a total of 27 studies from 2003-2019. The DNA methylation meta-analysis is based on a total of 15 studies from 2000-2019. Significant association were found between telomere length and DNA methylation and all-cause mortality, suggesting an important role of epigenetic markers and aging process.
Materials and Methods
The data for this project were from the PubMed database (https://www.ncbi.nlm.nih.gov/pubmed). Although mortality could be due to many causes, this study was limited to all-cause mortality. All-cause mortality does not correspond to death from a specific disease, but rather including many aging-related diseases, and can serve as a proxy to aging process [11]. The data for this project were primarily from a recent review published in 2018 [12], which included 23 studies meta-analyzed using a random-effect model. Four additional studies were found in 2018 and 2019 [13- 16]. One study (ESTHER) [14] was included in the previous metaanalysis, but the new publication expanded telomere measures from 3,566 to 9,638 participants. Therefore, the initial ESTHER results were replaced with the latest samples. When carefully examining details of individual studies, heterogeneity was observed in telomere length measurement (quantitative polymerase chain reaction (qPCR) vs Southern blotting (SB)), coding of telomere length (e.g., continuous, tertiles, quartiles, quintiles), statistical model (e.g., logistic regression, cox regression, and Mann-Whitney U test), and confounder adjustment. The individual study results were not directly comparable, and a pooled estimate for the effect size was not feasible.
In this analysis, a weighted z-score meta-analysis was used, which weighted the z-statistic from each individual study by their sample size (number of deaths). This method does not combine the effect size estimates but used z-statistic as measure of association strength between telomere length and all-cause mortality. A similar search was conducted on the PubMed database to search for publications on DNA methylation markers and allcause mortality, using the keywords “epigenetic” and “all-cause mortality”. A recent meta-analysis was found using 12 cohorts in a collaborative approach [17], where the involved research groups agreed to perform association tests between DNA methylation age and all-cause mortality using consistent modeling and share the results, either negative or positive. Three additional studies were carried out separately. Results from these 15 cohorts was combined using both fixed- and random-effect meta-analysis. Multiple formulas have been developed, based on different sets of methylation markers, to calculate “DNA methylation age”. It is typically compared to chronological age to obtain a measure of age acceleration. A formula derived by Horvath [18] was selected for the meta-analysis as much as possible (14 out of 15 cohorts).
Data
We introduce telomere length data and DNA methylation data that are used in the study, respectively.
Telomore Length Data
A total of 27 studies were included in the meta-analysis that reported association results between telomere length and all-cause mortality. Summary characteristics of individual studies were shown in Table 1. Individual association results were included in Figure 1, except Igari, et al. [16], which only reported association p-value instead of estimated effect size (hazard ratio, HR). When effect sizes (HRs) and their 95% confidence intervals (CIs) were available, we first converted the HR and CI into natural log, and then calculated z-score as log(HR)/SE(HR), where the standard error (SE) was estimated from log-transformed CI. The p-value reported in Igari, et al. [16] was converted into z-score from the inversequantile function of standard normal distribution.
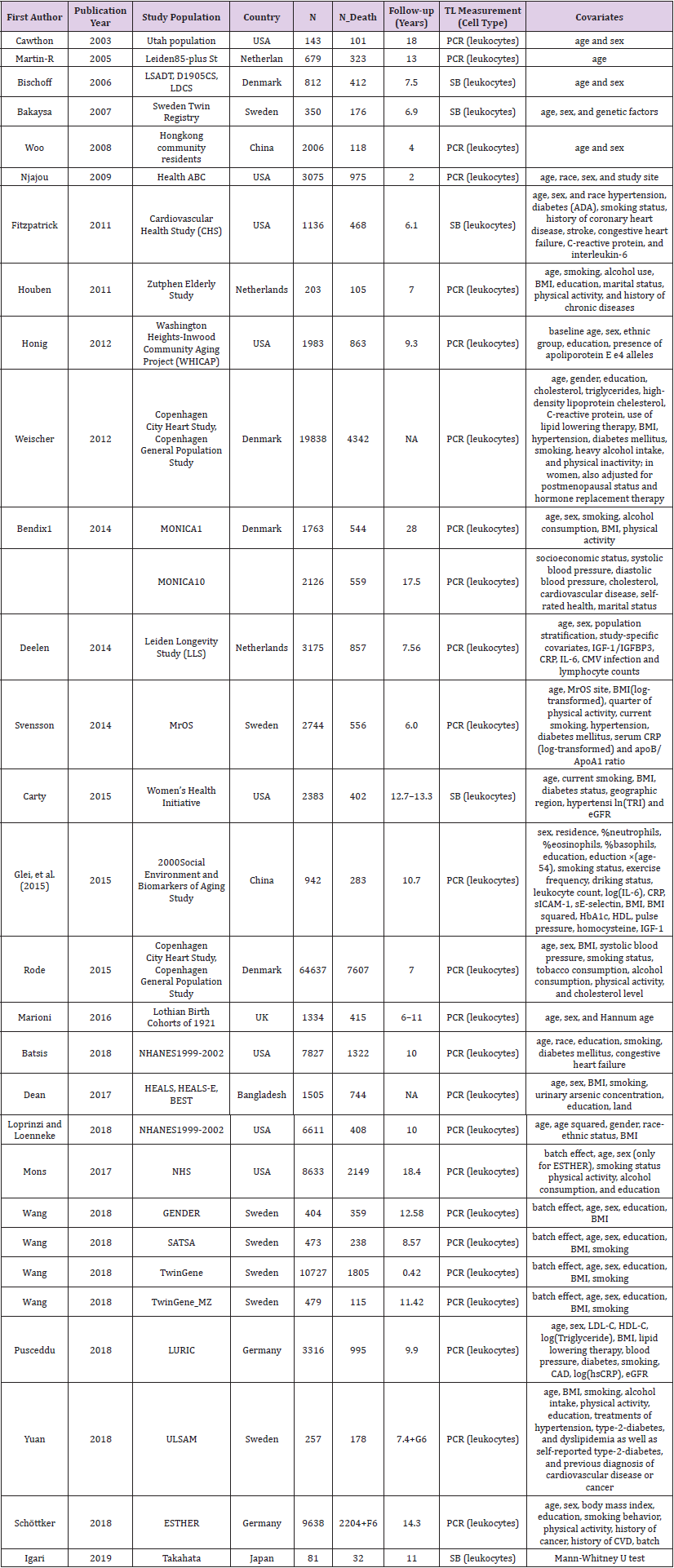
Table 1: Telomere length data.
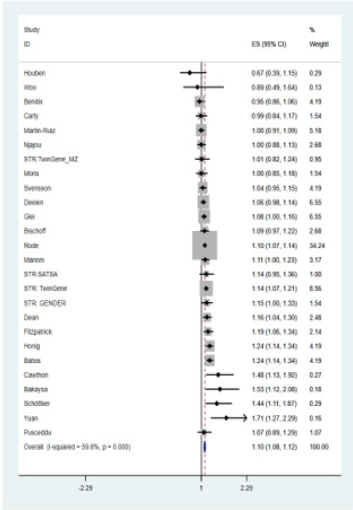
Figure 1: Forest plot of telomere length and all-cause mortality.
DNA Methylation Data
A total of 15 studies were included in the meta-analysis that reported association results between DNA methylation and allcause mortality. Summary characteristics of individual studies were shown in Table 2. There were a total of 16,939 participants with 3,634 deaths. Most of the studies were on whites, except two on blacks and one on Hispanic. Results from these 15 cohorts was combined using both fixed- and random-effect meta-analysis. Multiple formulas have been developed, based on different sets of methylation markers, to calculate “DNA methylation age”. It is typically compared to chronological age to obtain a measure of age acceleration. A formula derived by Horvath [18] was selected for the meta-analysis as much as possible (14 out of 15 cohorts).
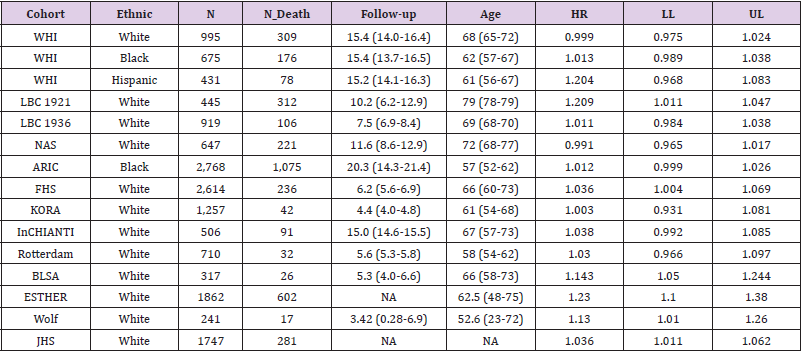
Table 2: DNA methylation data.
Results
We report telomere length results and DNA methylation results, respectively.
Telomore Length Results
Twenty-six of the 27 studies reported estimated effect size as odds ratio or hazard ratio between reduced telomere length and all-cause mortality. Given heterogeneity among these studies, a pooled estimate did not have a direct interpretation. A forest plot (Figure 1) was generated to show individual association results, but only for demonstration purpose. A weighted z-score method was used for meta-analysis. A significant association was observed between telomere length and all-cause mortality (combined z = 7.49, p = 6.75E-14).
We further restricted analysis into different ethnic groups. There were 23 studies conducted in European countries or United States. The combined z-score was 6.85 (p = 7.34E-12). Another 3 studies were based on Eastern Asian population (specifically, China and Japan). Although the sample sizes were much smaller in these 3 studies compared to many Europe- or US-based studies, their results still yielded a combined z-score of 3.56 (p-value = 0.0004). Therefore, the telomere length was significantly associated with all-cause mortality in both of the two ethnic groups. Although not appropriate due to the inconsistent effect size estimates, we still applied a funnel analysis to diagnosis potential publication bias, in which negative/insignificant findings would be less likely to publish.
The funnel plot (Figure 2) did not suggest any obvious publication bias, with most of the studies being inside of the confidence interval (the “funnel”).
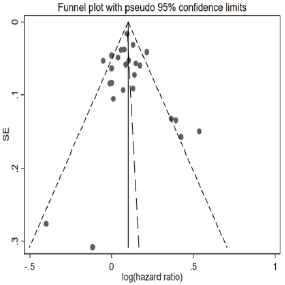
Figure 2: Funnel plot for potential publication bias.
DNA Methylation Results
Results from 15 independent cohorts (2014-2019) were combined first using fixed-effects meta-analysis. However, moderate heterogeneity has been observed among the results (Figure 3, heterogeneity test p-value = 0.022). A random-effect meta-analysis was therefore carried out to account for this heterogeneity. A combined effect size was estimated for 2.2% higher risk of death per 1-year age acceleration (DNA methylation age minus chronological age) (z-statistic = 153.3, p < 1E-308). As most of the studies (12 out 15) were conducted in a collaborative manner, where both negative and positive results were shared, significant publication bias was not expected. When we limited our analysis to whites only (12 studies), we still observed significant p-value for the heterogeneity test (p-value = 0.002). Random-effect meta-analysis gave an estimate of 2.1% higher risk per 1-year age acceleration (z-statistic = 109.6, p < 1E-308).

Figure 3: Forest Plot for DNA Methylation Age Acceleration and All-Cause Mortality.
Conclusion and Discussion
In this project, we carried out an extensive literature review on epigenetic markers and ageing. Using a meta-analysis approach, we observed significant association between telomere length, a well-known epigenetic marker, and all-cause mortality as a proxy for ageing, from a total of 27 individual studies. Subgroup analysis also con-firmed the association in both European/US and Eastern Asian populations. Similar finding was observed between DNA methylation and mortality, using a meta-analysis of 15 individual cohorts. These suggest an important role of epigenetic markers and aging process. One limitation of the project was the lack of original data. Although meta-analysis is a power method to combine results from multiple studies, these results in our analysis were heterogeneous in terms of telomere length measurement (PCR vs SB), coding of variable (continuous vs categorical), and covariates controlled for, etc. We had to choose a less powerful statistical method (weighted z-score) instead of a random-/fixed-effects regression method to obtain robust results.
Besides telomere length, other epigenetic markers have also been reported for association with ageing. For example, Zhang, et al. identified significant association be-tween DNA methylation score and all-cause mortality. Schöttker, et al. [14] compared various epigenetic markers, including telomere length, DNA methylation predicted age, 8-isoprostane levels, and 25(OH)D levels, on their association with all-cause mortality. In their joint model of these epigenetic markers, telomere length showed significant association while DNA methylation predicted age did not. In contrast, Gao, et al. [19] showed significant association between DNA methylation age and all-cause mortality after controlling for telomere length. Because DNA methylation is prevalent on the DNA sequence with millions of methylation markers, it potentially provides a stronger prediction tool than telomere length for aging. DNA methylation data are also easily accessible through public databases, allowing more complicated modeling approaches than meta-analysis. My next step will focus on DNA methylation on its relationship with the ageing process.
Future Work
In this preliminary analysis, we confirmed association between telomere length and all-cause mortality. It suggests the role of epigenetic markers in the ageing process. In future study, we will explore other epigenetic markers, e.g., DNA methylation, and develop an efficient prediction model for ageing and age acceleration using data mining tools.
Data Availability Statement
The data presented in this study are searched from https:// www.ncbi.nlm.nih.gov/pubmed and is available upon request to the author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jaul E, Barron J (2017) Age-related diseases and clinical and public health implications for the 85 years old and over population. Frontiers Public Health 5: 335.
- Franceschi C, Garagnani P, Morsiani C, Conte M, Santoro A, et al. (2018) The continuum of aging and age-related diseases: com-mon mechanisms but different rates. Front Med 5: 61.
- Bocklandt S, Lin W, Sehl M, Sanchez F, Sinsheimer J, et al. (2011) Epigenetic predictor of age. PloS one 6(6): e14821.
- Jylhävä J, Pedersen N, Hägg S (2017) Biological age predictors. EBioMedicine 21(4): 29-36.
- Herrmann M, Pusceddu I, März W, Herrmann W (2018) Telomere biology and age0related diseases. Clinical Chemistry and Laboratory Medicine 56(8).
- Zgheib NK, Sleiman F, Nasreddine L, Nasrallah M, Nakhoul N, et al. (2018) Short telomere length is associated with aging, central obesity, poor sleep and hypertension in lebanese individuals. Aging Diseases 9(1): 77-89.
- Chahine MN, Toupance S, El Hakim S, Labat C, Gautier S, et al. (2019) Telomere length and age-dependent telomere attrition: the blood-and-muscle model. Canadian Journal of Physiology and Pharmacology 97(4).
- Phillips T (2008) The role of methylation in gene expression. Nature Education 1(1): 116.
- Hannum G, Guinney J, Zhao L, Hughes G, Sadda S, et al. (2012) Genome-wide methylation profiles reveal quantitative views of human aging rates. Molecular cell 49(2): 359-367.
- Levine M, Lu A, Quach A, Chen B, Assimes T, et al. (2018) An epigenetic biomarker of aging for lifespan and healthspan. Aging 10(4): 573-591.
- R Marioni, S Shah, A McRae, B Chen, E Colicino, et al. (2015) DNA methylation age of blood predicts all-cause mortality in later life. Genome biology 16(1): 25.
- Wang Q, Zhan Y, Pedersen N, Fang F, Hägg S (2018) Telomere length and all-cause mortality: a meta-analysis. Ageing Research Reviews 48: 11-20 .
- Yuan X, Kronstrom M, Hellenius M, Cederholm T, Xu D, et al. (2018) Longitudinal changes in leukocyte telomere length and mortality in elderly Swedish men. Aging 10(10): 3005-3016.
- Schöttker B, Hagen L, Zhang Y, Gao X, Holleczek B, et al. (2018) Serum 25-hydroxyvitamin D levels as an aging marker: strong associations with age and all-Cause mortality independent from telomere length, epigenetic age acceleration, and 8-isoprostane levels. The Journals of Gerontology 74(1): 121-128.
- Pusceddu I, Kleber M, Delgado G, Herrmann W, Marz W, et al. (2018) Telomere length and mortality in the ludwigshafen risk and cardiovascular health study. PloS ONE 13(6): e0198373.
- Igari R, Davy P, Sato H, Takahashi Y, Iseki C, et al. (2019) Cognitive impairment, brain ischemia and shorter telomeres are predictors of mortality in the Japanese elderly: A 13-year prospective community-based study. Journal of The Neurological Sciences 397: 129-134.
- Zhang Y, Wilson R, Heiss J, Breitling LP, Saum KU, et al. (2017) DNA methylation signatures in peripheral blood strongly predict all-cause mortality. Nature Communications 8: 14617.
- Horvath S, Zhang Y, Langfelder P, Kahn R, Boks MPM, et al. (2012) Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biology 13(10): R97.
- Gao X, Zhang Y, Mons U, Brenner H (2018) Leukocyte telomere length and epigenetic-based mortality risk score: associations with all-cause mortality among older adults. Epigenetics 13(8): 846-857.
