 Case Report
Case ReportAbstract
Hereditary non-polyposis colorectal cancer (Lynch syndrome) is a multiorgan cancer syndrome, which is caused by an autosomal dominant mutation in germ line in the DNA mismatch repair (MMR) genes MLH1, MSH2, MSH6 and PMS2 [1]. The diagnosis and follow-up by expert oncologists play an important role in the screening and early diagnosis of tumors in these patients. The malignancies associated to MSH6 germline mutation include colorectal, endometrial, small bowel, urinary tract, and brain tumors. However, brain tumors represent a rare and relatively uncharacterized tumor type in Lynch syndrome [2].
Keywords: Glioblastoma; IDH- 1 Mutant; Mismatch Repair (MMR); Lynch Syndrome
Introduction
We present a 36-year-old female affected of ulcerative proctitis treated with rectal mesalazine and with no other pathological previous history. Her father had been diagnosed of Lynch Syndrome with germinal mutation in MSH6 c.3850dupA (p.Thr1284AnsfsX5).
Clinical Summary
In October 2012, the patient presented partial seizures and two months later, a rapidly progressive motor dysphasia was established. In February 2013, a cranial magnetic resonance imaging (MRI) showed a left frontotemporal contrast-enhancing lesion with associated edema. Levetiracetam 500 mg twice a day was initiated with control of the seizures and on February 28th, 2013 a macroscopic complete resection was carried out with favorable evolution after surgery. Radiological evolution is showed in (Figure 1).
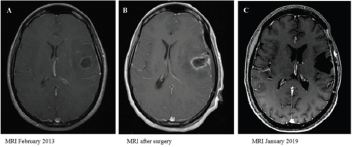
Figure 1:
A. T1-weighted post-gadolinium MRI pre-surgery;
B. post-surgery and
C. last study after a progression-free interval of 6 years.
Pathological Findings
Most of the tumor cells were gemistocytic neoplastic astrocytes, with globose, well-defined eosinophilic cytoplasm with displacement of the nucleus to the periphery (Figure 2). The mitotic index was 2 mitosis per 10HPF. Some vessels with endothelial proliferation were observed. There were no foci of necrosis. In the immunohistochemical study, the tumor cells were positive for IDH- 1 (anti-IDH1 R132H), and p53 (more than 70% of tumor cells) and they presented loss of ATRX nuclear expression. The MIB-1 index was 3%. Immunohistochemical analysis of MSH6 protein showed absence of nuclear staining of tumor cells. The histologic diagnosis was Glioblastoma, WHO grade IV, IDH1-mutated. Next generation sequencing (NGS) panel (Foundation Medicine) was performed in DNA isolated from FFPE tumor tissue. The Foundation Medicine T7 test assay interrogates 395 genes for base substitutions, insertiondeletions and copy number changes. Histological image and NGS results are presented in (Figure 2). Due to the anatomopathological diagnosis of IDH-1 mutated and ATRX mutated glioblastoma standard treatment with Stupp protocol was initiated. Patient received chemoradiotherapy with total radiotherapy dose of 60 Grays administered in 30 sessions with concurrent daily temozolomide from April 17th to June 6th , 2013 followed by six cycles of temozolomide. Follow-up was afterwards initiated with periodic MRI without any evidence of tumor relapse. In fact, there is no evidence of disease in the last MRI performed in January 2019.
In October 2015 genetic diagnosis was performed due to family history of MSH6 mutation and positive MSH6 mutation in tumor sample detected with NGS (Foundation Medicine Panel). This result was confirmed with the detection of mutated MSH6 p.Thr1284AsnfsX5 in patient’s germinal line and also with the absence of MSH6 immunohistochemical staining in the tumor cells (Figure 2).
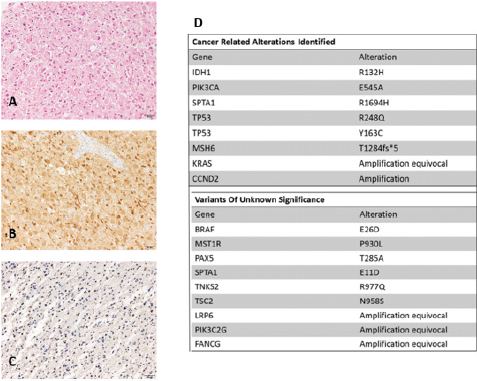
Figure 2:
A. Resection tissue shows conspicuous proportion of gemistocytic neoplastic astrocytes (hematoxylin & eosin staining)
B. Tumor cells are positive for mutant IDH1 (anti-IDH1 R132H)
C. Immunohistochemical analysis of MSH6 shows absence of nuclear staining of tumor cells
D. Foundation Medicine panel results.
Discussion
Some efforts have been made to understand tumorigenesis of different tumors in Lynch syndrome [3]. Nowadays it is not clearly understood why certain locations are more susceptible than others to develop cancer in the context of deficient MMR. Lynch syndrome. It has been estimated to confer a 1%–6% risk of brain tumors [4]. In each tumor location, it seems to occur different molecular changes after predisposing MMR deficiency. To date, association between MSH6 mutation and glioblastoma drug resistance to temozolomide has been briefly described. Small series of cases showed that somatic inactivation of MSH6 is reported in recurrent glioblastoma treated with alkylating agents but not in pre-treatment samples [5]. Additionally, MSH6 mutations are associated with resistance to chemotherapy in recurrent GBM [5]. Regarding Lynch syndrome, brain tumors occurred in 14% of the families and all glioblastoma patients described so far were IDH wt [6]. The peculiarity of this case consists in the favorable clinical evolution and its molecular profile with IDH1 mutation. In fact, the patient is currently asymptomatic and disease-free after 6 years since diagnosis. We hypothesize that the phenotype associated with IDH1 mutation prevails over the hypermutated phenotype linked to MSH6 mutation. A matter to be discussed is if the IDH1 mutation was a consequence of the MMR deficiency.
Acknowledgement
“This work was supported by the Instituto de Investigación Carlos III - Ministerio de Economía y Competitividad de España: Acción Estratégica en salud fund PI17/01621 to JMS-S.”
Disclosures
Authors included in this case report declare that they have no conflict of interest.
References
- Abdel Rahman WM (2006) The genetics of HNPCC: Application to diagnosis and screening. Crit Rev Oncol Hematol 58: 208-220.
- Ilencikova D, Sejnova D, Jindrova J, Babal P (2011) High grade brain tumors in siblings with biallelic MSH6 mutations. Pediatr Blood Cancer 57(6): 1067-1070.
- AHS Gylling, TT Nieminen, WM Abdel Rahman, K Nuorva, M Juhola, et al. (2008) Differential cáncer predisposition in Lynch Syndrome: Insights for molecular analysis of brain and urinary tract tumors. Carcinogenesis 29(7): 1351-1359.
- Barrow E, Robinson L, Alduaij W, A Shenton, T Clancy, et al. (2009) Cumulative lifetime incidence of extracolonic cancers in Lynch syndrome: A report of 121 families with proven mutations. Clin Genet 75: 141-149.
- Yip S, Miao J, Cahill DP, Iafrate AJ, Aldape K, et al. (2009) MSH6 mutations arise in glioblastomas during temozolomide therapy and mediate temozolomide resistance. Clin Cancer Res 15(14): 4622-4629.
- Therkildsen C, Ladelund S, Rambech E, Persson A, Petersen A, et al. (2015) Glioblastomas, astrocytomas and oligodendrogliomas linked to Lynch síndrome. Eur J Neurol 22(4): 717-724.
