 Review Article
Review ArticleAbstract
The Virus, Variants and Pandemic: COVID-19 as a disease and SARS-CoV-2 as its causative organism, continue to remain an enigma. The pandemic continues to persist over a year with outbreaks and re-emergence involving newer regions and population groups. While we continue to explore the agent factors, disease transmission dynamics, pathogenesis and clinical spectrum of the disease, and therapeutic and vaccine modalities, the grievous nature of the disease is evolving with the genomic mutations and pathophysiological alterations.
The Emerging SARS-Cov-2 Variants: Understanding the nucleotide variations in the virus genome provides a useful insight for the changing face of the disease and propagation of the pandemic. The emerging mutations can be interpreted as a strategy through natural selection to facilitate extensive spread of the viral infection. Of late, there is recurrent emergence, accumulation, and onward transmission of various mutations. Among the current major SARS-CoV-2 lineages A, B, B.1, B.1.1, and B.1.177, the lineage - B.1.1.7 is of present concern.
Challenges Posed by the Mutations: In general, the D614G mutation leads to open conformations, increases density of the S protein and disease infectivity by facilitating the viral entry into the host cells. Another variant, named VUI-202012/01 or B.1.1.7 identified in the UK, harbours multiple mutations which have been correlated with a significantly increased infection transmission rate during recent months. A new entrant, the South African variant carries a high infectivity and accompanied by probable high disease severity. There are salient potential consequences of emerging variants including rapid transmission, enhanced disease severity, and diagnostic failure as the variants may evade detection through certain diagnostic tests. Further, they may decrease susceptibility to antiviral drugs and monoclonal antibodies and evade natural or vaccineinduced immunity.
Conclusion: Dealing with the Variants: The WHO is working with various countries to amplify and adapt the current surveillance systems to evaluate potential virus variations through ongoing systematic clinical and epidemiologic surveillance. There is need to establish genomic sequencing capacity and access to the sequencing services. Simultaneously, the disease control measures, and intensive public health response are needed to be strengthened to curtail the increased transmissibility associated with the emerging SARS-CoV-2 variants.
Keywords: Clades and lineages; Danish Cluster 5 Variant; Escape Variants; Neutralising Antibodies; Receptor Binding Domain; Spike Protein; South African Variant; B.1.1.7; D614G; VUI-202012/01, 5O1Y.V2 Variant
Introduction
The Virus and Pandemic
COVID-19 as a disease and SARS-CoV-2 as its causative organism, continue to remain an enigma. The Covid-19 pandemic continues to persist over a year with outbreaks and re-emergence and involvement of newer regions and population groups. COVID-19 has changed the ways of life for mankind, damaged economies, and altered sociocultural structure. Moreover, it appears that the SARS-CoV-2 virus is here to stay for the foreseeable future [1] and continue to create further deterioration and havocs.
Mutations and Variants of SARS-CoV-2
While we continue to explore the agent factors, disease transmission dynamics, pathogenesis and clinical spectrum of the disease, and therapeutic modalities, the grievous nature of the disease is evolving further with the genomic changes in the virus and pathophysiological alterations and clinical manifestation. The future of the disease, of course, is stated to depend on various known and unknown factors, which include the effects of seasonality on the disease transmission and spread. At the individual level, it is to depend on the degree and duration of effective immune response to the disease, and at the cohort and societal level on the lifestyle choices made by individuals and measures enforced by the governments. Further, the pandemic’s course will also depend on the availability of a COVID-19 vaccine, its efficacy and duration of its protective immune response. Concerning the latter, the significance of viral mutations should also be highlighted and explored further. There are serious challenges posed by SARS-CoV-2 virus and COVID-19 as the disease, that are further increased by mutations and evolution of variants with enhanced infectivity and probably virulence. Looking forward, let us hope that the today’s unmet challenges are resolved in near future [2].
The Viral Variants Vs Strains
Being an RNA virus, SARS-CoV-2 has a steady rate of mutations despite presence of the spell-checker, nsp14 protein acting as 3′-5′ exoribonuclease. It is expected and has been observed that the virus accumulates mutations over time. With the mutations, the variants may lose or gain infectivity and virulence. As feared, enhancing the infectivity and virulence due to mutations may pose a heightened challenge for Covid-19 therapeutic and preventive modalities, including vaccines. There is no universally accepted definition for the terms strain, or variant. According to van Regenmortel et al a virus strain is a variant of a given virus that is recognizable because it possesses some unique phenotypic characteristics that remain stable under natural conditions [3]. Whereas the unique phenotypic characteristics and biological properties for a virus strain are different from the compared reference virus, such as the antigenic properties, host range or the disease manifestations it can cause [4]. Further, a virus variant with a simple difference in genome sequence should not be given the status of a separate strain since it lacks a distinct recognizable viral phenotype [5]. Mutations in SARS-CoV-2 are common and over 4,000 mutations have been detected in the spike (S) glycoprotein alone, according to the observations by COVID-19 Genomics UK (COG-UK) Consortium.
SARS-CoV-2 Clades and Lineages
There have been identified several thousand variants of SARSCoV- 2. These variants have been placed under larger groups called clades. Due to lack of uniformity, there have been proposed several nomenclatures for SARS-CoV-2 clades. As of December 2020, the Global Initiative on Sharing All Influenza Data (GISAID) in reference to SARS-CoV-2 or hCoV-19, has identified seven clades - O, S, L, V, G, GH, and GR [6]. Whereas Nextstrain has, as of December 2020, found five - 19A, 19B, 20A, 20B, and 20C [7]. In this reference, Guan et al. have recently named five global clades - G614, S84, V251, I378 and D392 (8). Further, Rambaut et al. have proposed the term ‘lineage’ and identified five major lineages A, B, B.1, B.1.1, and B.1.177 [8]. Both the Nextstrain and the GISAID clade nomenclatures, in general, aim at a broad categorization of globally circulating diverse SARSCoV- 2 variants, whereas the lineages nomenclature by Rambaut et al has been related to the disease outbreaks and re-emergence.
Trying to simplify the issue, a WHO document has identified six major clades with 14 subclades [9,10]. The largest clade is D614G clade with five subclades. Within D614G clade, D614G/Q57H/T265I subclade forms the largest subclade with 2391 samples. The second largest major clade is L84S clade, observed among travellers from Wuhan and consists of 1662 samples with 2 subclades. The L84S/ P5828L/ subclade is predominantly observed in the United States. Whereas G251V frequently appears in samples from the United Kingdom, Australia, the United States, and Iceland. The remaining two clades D448del and G392D are smaller and without substantial subclades at this point.
Emerging SARS-CoV-2 Variants
Mutations and Variation in SARS-CoV-2
Understanding the nucleotide variations in the SARS-CoV-2 genome provides a useful insight for evolution of the disease and propagation of the pandemic [11]. The early variations have made their way almost unnoticed as the virus spread around the world. Whereas most variations or mutations have no impact on the viral ability to transmit or cause disease, certain mutations appear to have impact on transmissibility, infectivity, or lethality. Some of these mutations have possibly arisen as a result of the virus evolving from immune selection pressure in infected individuals and are more prevalent in patients with mild than those with severe disease. In general, the mutations can be interpreted as a strategy through natural selection to facilitate extensive spread of the viral infection. As such the SARS-CoV-2 virus has a low mutation rate by virtue of the nsp14 protein acting as 3′-5′ exoribonuclease on both single-stranded and double-stranded RNA during the viral replication cycle [12]. Still its large genome appears to facilitate recombination, insertions, and deletions. Andrés et al have noted that the viral S protein accumulates deletions upstream and close to the S1/S2 cleavage site [13]. Further, SARS-CoV-2 can resort to RNA viral evolution through recombination (synthesis of chimeric RNA molecules from two different progeny genomes) and reassortment (the packaging within a single virion of genomic segments from different progeny viruses).
In general, the Single Nucleotide Variations (SNVs) as SARSCoV- 2 Spike amino acid replacements in the Receptor Binding Domain (RBD) occur relatively frequently [14]. There is recurrent emergence and significant onward transmission of a six-nucleotide deletion in the S gene resulting in loss of two amino acids labelled as ΔH69/ΔV70. This deletion often co-occurs with the receptor binding motif amino acid replacements N501Y, N439K and Y453F. As such, these deletions have been found in a small percentage (2.2%) of the samples [15]. Among the Current major SARS-CoV-2 Lineages A, B, B.1, B.1.1, and B.1.177, the lineage - B.1.1.7, of present concern. First sequenced in the UK on 20 Sep 2020, it is spreading to other countries and has been discovered in Denmark, the Netherlands, Italy, Israel, Australia, Hong Kong, Singapore, Japan, and the USA. The other countries are being increasingly involved. Using the complete sequences of 1,932 SARS-CoV-2 genomes, six types of the variants have been identified. The 13 signature variations in the form of SNVs in protein coding regions and one SNV in the 5′ untranslated region (UTR) provide interpretation for the six types (types I to VI). The type VI, characterized by the four signature SNVs C241T (5′UTR), C3037T (nsp3 F924F), C14408T (nsp12 P4715L), and A23403G (Spike D614G), with strong allelic associations, first reported in China, has become the dominant type world over.
Out of these, C241T is in the 5′ UTR appears to be of uncertain significance. The other three SNVs, 3037T-14408T-23403G characterising the increasing frequency of the type VI, in majority of samples from various regions indicate a possible fitness gain for the virus. Further, it has been noted that the variants missing one or two of these signature SNVs fail to persist or wiped out by other evolutionary more fit variants [16]. Taking a note of the major mutations, their lineages and effects on disease transmissibility is important to understand the changing face of the pandemic (Table 1).
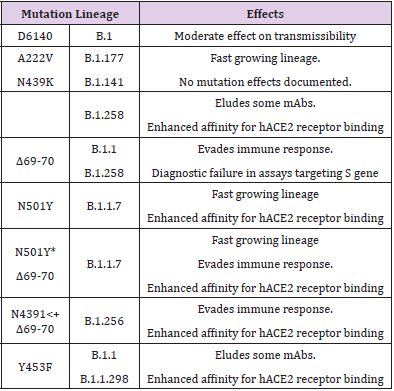
Table 1: Major mutations, lineages and effects on disease transmissibility and course.
Emergence of the D614G Variant
The genomic analysis of various samples for SARS-CoV-2 from several regions has found an increased proportion of some particular variants. One such variant is the D614G mutation in the C-terminal end of the S1 domain and in proximity to the S2 subunit. In short called the G variant, it has increased in prevalence during the pandemic, probably after initially arising in China and then spreading to Italy in January and later globally to become the dominant form in the pandemic [17]. The SARS-CoV-2 G variant is part of the G clade by GISAID and the B1 clade by the Phylogenetic Assignment of Named Global Outbreak LINeages (PANGOLIN). The variant is associated with the faster viral transmission and harbouring and discharge of higher viral loads by virtue of higher binding to the ACE2 receptor and higher protein stability [18]. In addition, it is associated with reduced S1 Shedding. The presence of glycine at 614, thus, appears to improve the S-Protein Stability and Increase its incorporation into virions. Further, the structural analyses have revealed that the RBD of the D614G form of the spike protein is more likely to assume an ‘open’ conformation than the RBD of the ancestral D614S form, resulting in an improved ability to bind to the hACE2 receptor. Furthermore, the higher viral load with D614G is consistent with epidemiological data suggesting enhanced infectivity associated with D614G [19].
Studies in human respiratory cells and in animal models demonstrated that compared to the ancestral virus, the variant with the D614G substitution has increased infectivity and transmission. With the studies in hamsters infected with D614S and D614G variants, Plante et al. have documented that the D614G variant replicated to higher titers in nasal-wash samples early after infection than the ancestral D614S variant. These findings suggest increased viral fitness for D614G in the major upper airway compartment potentially associated with enhanced transmission [20]. Experimentally, the D614G variant was found to be equally sensitive to neutralizing antibodies and did not cause more severe disease than the ancestral variant in hamsters, an observation supporting current findings in humans [21]. As shown, the SARSCoV- 2 virus with the D614G mutation is more infective in laboratory tests. But the results of lab tests cannot be directly extrapolated to the real-world occurrence as a lot of additional factors are involved in human transmission, including host-pathogen interactions, host genetics and other epidemiological considerations. Further, the mutation does not appear to have impact on the virulence and the SARS-CoV-2 virus with the D614G substitution does not cause more severe illness or alter the effectiveness of existing laboratory diagnostics, therapeutics, vaccines, or public health preventive measures.
In general, the D614G mutation leads to more open RBD conformations, increases density of the S protein and disease infectivity by facilitating the viral entry into the host cells [22]. Further, this mutation is often accompanied by other mutations involving parts of the SARS-CoV-2 genome. Epidemiologically, the rapid spread of D614G was first spotted in early samples collected from China and Germany. In due course, it has become the dominant strain across the European continent, Australia, Canada, and parts of the United States, and probably rest of the world regions. It appears that D614G represents a more transmissible form of SARSCoV- 2, which has emerged as a product of natural selection [18]. There is a potential concern that a similar situation may occur with the VUI-202012/01 or B.1.1.7 variant.
Emergence of VUI-202012/01 Variant
Another variant, named VUI-202012/01 (Variant Under Investigation, year 2020, month 12, variant 01) or the phylogenetic cluster named lineage B.1.1.7 has been identified through viral genomic sequencing in the UK. It has 17 mutations that may lead to a conformational change in the shape of the virus including the S protein. Out of the acquired 23 changes, there are 14 nonsynonymous (Amino Acid [AA] altering) mutations, 6 synonymous (non-AA altering), and 3 deletions. The deletions include a double 69/70 deletion and P681H - near the S1/S2 furin cleavage site, in the S protein, and ORF8 stop codon (Q27stop) - in open reading frame 8. Apart from this, there are multiple S protein mutations such as N501Y, A570D, D614G, Y453F, T716I, S982A, D1118H as well as mutations in other genomic regions. Recently a distinct phylogenetic cluster (named lineage B.1.1.7) was detected within the COG-UK surveillance dataset. This cluster accounts for an increasing proportion of cases in various parts of England [9,23]. Further, the preliminary analyses in the UK suggests that the UK variant B.1.1.7 having multiple mutations is significantly more transmissible than previously circulating variants, with an estimated potential to increase the reproductive number (R) by 0.4 or greater with an estimated increased transmissibility of up to 70 percent [24]. As reported by the UK, there is no clear epidemiological link to animals for VUI 202012/01 [25].
The variant was first detected during October 2020 in the UK from a sample taken in Sep 2020. The backwards tracing using genetic evidence suggests this new variant first emerged in September 2020 and then circulated at low levels in the population until mid-November, and thereafter it has spread alarmingly by mid-December. It has been correlated with a significantly increased infection transmission rate in the UK, having a ‘selection coefficient’ of 0.70 (70%), with a generational interval of 6.5 days, and being 74% more transmissible [26]. The cases with the new variant have been detected from various European countries including Denmark, Netherlands, and Belgium. The impact of suddenly increasing spread of the VOC 202012/01 variant was not made out till late-December [27].
The emergence of VUI-202012/01 has certain likely implications:
a) There is a probability of a wider spread of the new virus variant across the European nations and continents.
b) The potential impact on SARS-CoV-2 diagnostics - The UK reports that the deletion 69-70 in the spike protein of the variant causes a negative result from S-gene RT-PCR assays applied in some laboratories in the UK. This specific mutation has occurred many times in different countries and is geographically widespread.
c) The potential impact on severity of disease in a population or groups - The available information regarding severity of the new virus variant is limited. To date, there is no indication of increased disease severity observed related to the variant, but the assessment is presumptive as most of the reported cases are people under 60 years old, who are less likely to develop severe symptoms.
d) Rapid increase of a SARS-CoV-2 variant with multiple spike protein mutations in the UK, endorses significantly increased infectivity. None of the previously described SARSCoV- 2 variants have shown this property.
e) Frequency of reinfections - The mutations observed in the new variant are related to the RBD and other surface structures, which may alter the antigenic properties of the virus. Based on this observation, it is likely that reduced neutralisation by antibodies may be seen. But so far there is no evidence for increased risk for reinfection.
f) The potential impact on vaccine effectiveness is a worrisome issue. It is being speculated based on limited studies that the efficacy COVID-19 vaccines may remain like the ancestral SSARS-CoV-2 virus.
The VOC-202012/01 variant, also known as lineage B.1.1.7, is notable for a high number of mutations it contains. In all it involves 23 mutations: 13 non-synonymous mutations, 4 deletions, and 6 synonymous mutations [9]. Whereas 17 mutations alter proteins, the remaining six do not. The most important change in VOC- 202012/01 is N501Y, the change from asparagine (N) to tyrosine (Y) at amino-acid position 501, inside the Receptor-Binding Motif (RBM) part of the RBD [28]. Another feature in the variant, the Q27stop mutation renders the ORF8 protein inactive, which is an immunoglobulin (Ig)–like protein modulating the pathogenesis by mediating Major Histocompatibility Complex I (MHC-I) degradation and suppressing type I interferon (IFN)–mediated antiviral response and associated to milder disease and better prognosis.
As such the mutations in the RBD can change antibody recognition, ACE2 binding specificity, and the infectivity. Thus, the mutation N501Y affects the receptor binding affinity of the spike protein and alone or in combination with the deletion at 69/70 in the N Terminal Domain (NTD) enhances the transmissibility. An examination of the global GISAID SARS-COV-2 sequence database shows that this N501Y mutation has been circulating sporadically earlier outside the UK, in Brazil in April, Australia in June-July, and in USA in July 2020 [29]. The same mutation being independently selected several times appears to imply its fitness for the virus. Presently the preliminary epidemiologic indicators suggest the B.1.1.7 variant is associated with higher viral loads in respiratory tracts and increased transmissibility associated at least partly with the N501Y mutation [30]. However, there is no indication of change in the disease severity as measured by length of hospitalization and 28-day case fatality. But on a careful note, a clinical study has documented that the variant in a patient was less susceptible to convalescent plasma than wild-type virus [14]. Thus, there are potential concerns that the B.1.1.7 variant might lead to more severe disease or even evade vaccine-induced immunity. The B.1.1.7 variant is reckoned to have spread in the UK following its probable emergence during September 2020 and rapidly replaced other variants, and since then has been detected in numerous countries around the world, including the US, Canada, Australia, and Japan.
The South Africa 5O1Y.V2 Variant
Another new variant, the South African lineage, has been detected as rapidly spreading in the Eastern Cape, Western Cape, and KwaZulu-Natal provinces in South Africa. This variant was first identified in Nelson Mandela Bay, South Africa, in samples dating back to the beginning of October 2020, and cases have since been detected outside of South Africa including Zambia in late December 2020. The variant bears the N501Y mutation and accompanied with a high viral load and increased transmissibility and has largely replaced other SARS-CoV-2 variants in the region. Currently, there is no evidence of this variant being associated with more severe disease or worse outcomes. The variant has additional mutations, E484K and K417N, in the RBD in the S glycoprotein and is associated with immune escape [31]. This variant named 20C/501Y.V2 or B.1.351 lineage has emerged independently of the B.1.1.7 lineage and shares some mutations with the B.1.1.7 lineage. It has multiple mutations in the spike protein, including N501Y but does not contain the deletion at 69/70. It is feared that out of the two N501R variants, V1 – the English and V2 - the South African, the latter with 2 other mutations, K417N and E484K, in the RBD of the S protein could be more infectious. The variant has been shown to infect younger people with no underlying health conditions and may resist the vaccine [32]. While the variants D614G, 202012/01 and 501Y.V2, and other variants appear to influence the disease infectivity, they do not appear to cause changes in clinical presentation or severity (Table 2). However, the higher case incidence may lead to an increase in COVID-19 hospitalizations and deaths and more intensive public health measures may be required to control transmission of these variants.
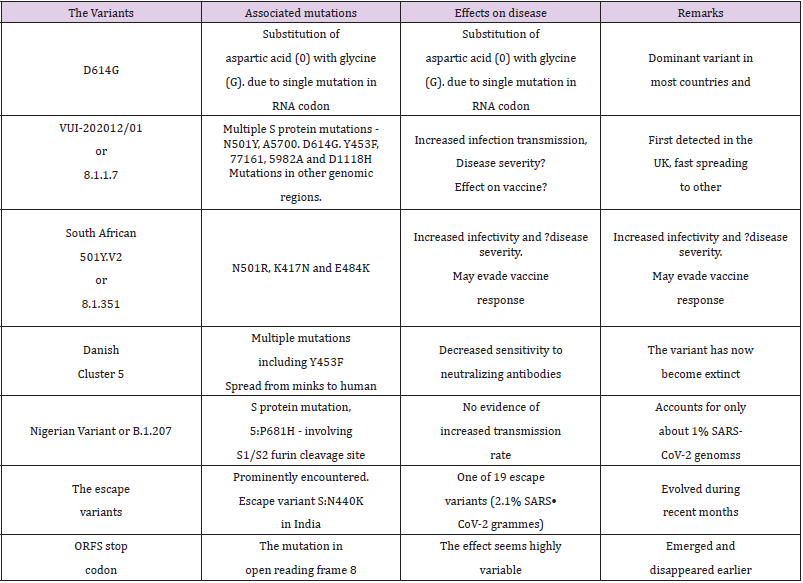
Table 2: Variants associated with concern and the variants to be watched.
The Danish “Cluster 5” Variant
The Cluster 5 variant, also referred to as ΔFVI-spike, was identified by the Danish State Serum Institute (SSI) during August 2020. It was discovered in Northern Jutland, Denmark, and is believed to have been spread from minks to humans in mink farms. The variant has multiple mutations and preliminary studies indicated that the variant may potentially result in reduced virus neutralization in humans, leading to decreased extent and duration of immune protection following natural infection or vaccination. As per the WHO communique also, the cluster 5 has been found to have a moderately decreased sensitivity to neutralizing antibodies [33]. Following identification, extensive investigation and surveillance, Danish authorities have identified only 12 human cases of the Cluster 5 variant in September 2020. Subsequent to culling of the mink population, lockdown. and travel restrictions, and masstesting the variant was held from spreading further, and SSI announced on 19 November 2020 that cluster 5 in all probability had become extinct [34].
The New Brazilian Variant
The new variant, also called P.1, belongs to the B.1.1.248 lineage of the coronavirus and as per the Japanese National Institute of Infectious Diseases (NIID) has 12 mutations, including N501Y and E484K, in its spike protein. Further research has shown that E484K could be associated with escape from neutralizing antibodies and may be able to evade parts of the body’s natural defence memory that bestows immunity. It is being conjectured that the P.1 variant has been circulating in Brazil’s Amazonas state since July of last year, before being detected in Japan during January 2021. The variant has high transmissibility and by eluding the immune response may be associated with re-infection.
Other Isolated and Minor Mutations
B.1.207 Variant: The P681H, a S protein mutation involving the S1/S2 furin cleavage site, has been recently reported from Nigeria. The P681H mutation is shared in VOC-202012/01, but has emerged spontaneously several times earlier also, and there is no evidence to indicate it contributing to increased transmission of the virus in Nigeria [35]. Earlier the analysis of samples collected in Aug 2020 and sequenced at the African Centre of Excellence for Genomics of Infectious Diseases (ACEGID), Nigeria, identified two SARS-CoV-2 sequences that share one non-synonymous SNP in S protein in common with this lineage. At the moment, only about 1% of the SARS-CoV-2 genomes from Nigeria share any of the 17 protein-altering variants from the UK lineage of concern (B.1.1.7) and other mutations such as the N501Y, A570D, and the HV 69 - 70 deletion in S protein have not been detected in Nigeria.
ORF8 Stop Codon (Q27stop): This mutation is not in the S protein but involves the open reading frame 8. The mutations akin to this, have occurred in the past. In Singapore, one strain with this type of mutation emerged and disappeared. This is in line with the understanding that SARS-CoV-2 ORF8 is poorly conserved among coronaviruses. Among accessory genes, open reading frame 8 (ORF8) stands out by being highly variable and may be related with the virus ability to spread [36].
S:N440K Variant: The CSIR Institute of Genomics and Integrative Biology (CSIR-IGIB) team in India has identified 19 genetic variants of the SARS-CoV-2 in India which have evolved to evade neutralising antibodies. Out of the 19 immune escape variants in India, one in particular known as S:N440K variant has been reported in 2.1 per cent of the gene sequences in India [37]. This variant appears to have evolved during the recent months. It has been associated with a case of reinfection in a 28-yearold female healthcare worker and possibly confers resistance to neutralising antibodies.
The Challenges Posed by Sars-Cov-2 Mutations
The Genomic Analysis of SARS-CoV-2 Variants
Of the 2969 missense variants, 1905 variants are found in ORF1ab, which is the longest ORF occupying two thirds of the entire genome. ORF1ab is transcribed into a multiprotein and subsequently cleaved into 16 Non-Structural Proteins (NSPs). Of these proteins, NSP3 has the largest number of missense variants among ORF1ab proteins. Of the NSP3 missense variants, A58T has been found to be the most common followed by P153L. The most common variants identified are the synonymous variant 3037C > T (6334 samples), ORF1ab P4715L (RdRp P323L; 6319 samples) and SD614G (6294 samples). Further, they are found to occur simultaneously in over 3000 samples, mainly from Europe and the United States [10]. The most common clade identified is the D614G variant, located in a B-cell epitope with a highly immunodominant region and may therefore affect vaccine effectiveness. Although amino acids are quite conserved in this epitope, there have been identified 14 other related variants [38].
Almost all variants with D614G mutation, in addition have a mutation in the protein responsible for replication (ORF1ab P4715L; RdRp P323L), which might affect replication speed of the virus. This protein being the target of the anti-viral drugs such as remdesivir and favipiravir, the probability of mutations may lead to emergence of the treatment resistant forms. Whereas the mutations in the RBD may not be selected further as they are likely to reduce receptor binding affinity which would decrease the virus fitness [39]. V483A and G476S are primarily observed in samples from the United States, whereas V367F is found in samples from China, Hong Kong, France, and the Netherlands. The V367F and D364Y variants have been reported to enhance the structural stability of the S protein and facilitate efficient receptor binding. All viruses, including SARS-CoV-2, change over time, most without a direct benefit to the virus in terms of increasing its infectiousness or transmissibility, and sometimes limiting propagation. The mutations alter RNA virus pathogenesis, virulence, transmissibility, or a combination of these.
The potential for virus mutation increases with the frequency of infections. Therefore, reducing transmission of SARS-CoV-2 by disease control measures as well as avoiding introductions to animal populations, are critical aspects to the global strategy to reduce the occurrence of mutations with negative public health implications. Further studies are required to understand the impact of specific mutations on viral properties and the effectiveness of diagnostics, therapeutics. and vaccines. Though, it is thought that a COVID-19 vaccine is likely to work, because vaccines use multiple targets to elicit an immune response [40]. The genome data outlines that two SARS-CoV-2 virus samples collected from anywhere in the world differ by an average of just 10 RNA letters out of 29,903 [41]. Thus, there is a sluggish mutation rate. But in spite of this, the researchers have catalogued more than 12,000 mutations in SARS-CoV-2 genomes. Most of the mutations appear not to affect the virus’s ability to spread or alter the disease severity. In fact, the mutations altering the viral structural proteins are more likely to harm a virus and make it extinct rather than improve it. On the other hand, an insignificant mutation can downgrade the immune response. Further, various mutations having no major impact might in future course of the disease and pandemic may accumulate to alter the infectivity and disease severity [42].
The COVID-19 pandemic in Africa and Asia
Till now the African countries have reported far fewer cases and deaths from COVID-19 than predicted. As of 22 November 2020, the continent of Africa, comprising 1.3 billion people, had recorded 2,070,953 cases of COVID-19 and 49,728 deaths [43]. This represents about 3.6% of total global cases and is far too low than the predicted up to 70 million Africans may be infected with SARS-CoV-2 with more than 3 million deaths by June 2020 [44]. A sero-survey study for measuring the occurrence of SARSCoV- 2 antibodies in blood donors in Kenya has highlighted that the incidence of SARS-CoV-2 infection is much higher than expected from case numbers [45]. Similarly, in October 2020, Mozambique reported less than 3000 confirmed cases of COVID-19; however, sero-surveys have found the actual transmission much higher [46]. This suggests that there may be more infections than documented. A similar scenario exists for other developing countries including India, where the Sero-survey study by the Department of Science and Technology (DST), has shown about 90 missed cases for every case detected. The DST has developed the Covid-19 India National supermodel indicating that 60% of the Indian population may have already been infected and recovered. The estimate was arrived based on mathematical and statistical model to map the trajectory of the virus and does not denote a failure of detection system but may indicate that the effect of the disease has not been severe [47].
The Ill-defined and Short-lived Immune Response
In the experimental studies using animals and cell cultures, along with the latest molecular techniques, and in small human clinical studies, the immune response to SARS-CoV-2 has been recognized [48]. Further, the animal studies in mice, primate studies in monkeys and human clinical studies have documented that those who received one of the experimental RNA vaccines, produced antibodies that proved more potent at blocking G viruses than D viruses [49]. This may be related to the more open conformation of its RBD. With the G variant now dominant globally, these findings are encouraging. So far, various SARS-CoV-2 variants do not stop the immune system from developing neutralising antibodies that recognize the virus. The researchers are, though, wary of the potential of SARS-CoV-2 to evade immunity and antigenic responses like the HIV. There exists a probability that the virus may acquire mutations that either evade the immune response or alter its susceptibility to antibodies and immune cells. Further, it is not certain whether the immunity will be effective or lasting. A related unanswered question is whether a vaccine will provide adequate protection, so that those who have recovered from COVID-19 or vaccinated can return to pre-pandemic behaviours. Furthermore, the reinfections have been documented to occur [50]. But so far neither the frequency nor the elements of the immune response associated with reinfection have been adequately researched and well documented.
The Effect of Mutations on COVID-19 Vaccines
Presently, the researchers as well as clinicians are most concerned about several mutations occurring in group in the S protein. The accumulation of multiple mutations as in the British variant or South African variant is more of a concern and could potentially impede immune protection. Another unresolved issue is the concern that the mutations can have far-reaching consequences for the human health in form of delayed disease complications. There is a chance that vaccines currently being administered in the country may not provide sufficient immunity against new variants emerging in both the UK and South Africa. Though, there is no evidence that the vaccines currently being administered will not be able to protect against these new variants. As such the mutations may make the virus less susceptible to the immune response triggered by the vaccines. It is being conjectured that even if the vaccine is not effective against the new variants, COVID-19 vaccine manufacturers might be able to make changes in the vaccines to add protection against the emerging variants. Scientists believe such changes, if needed can be made in about six weeks’ time.
The Staggered Covid-19 Vaccine Dosing in the UK
Facing rising infections and the upcoming ever-tightening lockdowns, the United Kingdom has reportedly decided to delay the second dose of both the Pfizer-BioNtech and Oxford-AstraZeneca vaccines. The proposed change from 21 days between doses to 12 weeks, will allow more people to get the protection of at least one dose [51]. There are concerns that the plan is different from the efficacy trials and may lead to unchartered course of immune response. In addition, the approach could foster vaccine-resistant forms and increase the potential for escape mutants by having so many people with incomplete protection into a community swamped with SARS-CoV-2 infections. There is another concern, the vaccination programme is covering the elderly citizens first, in whom the immune system does not function so well, and some may inevitably contract the disease while waiting for their second dose of vaccine which may also erode confidence in the vaccines.
The moves are borne of a desire to begin vaccinating as many people as quickly as possible. The moves are also based on evidence from ‘certain subsets of studies’ in participants in clinical trials, and on general principles of vaccinology rather than on actual research related to the specific vaccines being used [52]. The US-CDC has initially criticized the British move, though it appears that with the scarcity of enough vaccine doses in the immediate future, the US may also resort to such proposition, which may be followed by other countries around the world. The move has been endorsed by the WHO [53]. Further, the use of a non-matching second dose of vaccine is also disturbing. While there is reason to believe boosting with a different type of vaccine might be useful in some cases, particularly if the first dose is a vaccine like the AstraZeneca vaccine that uses a harmless virus onto which genetic material from SARS-2 has been fused. But the approach has not been studied in clinical trials and may lead to unfamiliar immune response.
Conclusion: Dealing with the Variants
The RBD and Non-RBD Mutations
The RBD of the S protein in SARS-CoV-2 plays a crucial role in binding with the hACE2 receptors required for viral entry. The intricate ACE2 receptor recognition mechanism of the SARS-CoV-2 virus regulates its infectivity and pathogenesis [54]. The mutations involving the RBD, thus, are associated with altered infectivity and possibly disease severity. On the other hand, the mutations distal to the RBD also impact the transmissibility and the antibody also appear to target non-RBD regions. A mutation in non-RBD region may lead to polybasic cleavage sites enhancement, via electrostatic interactions and hydration and influence the RBD-ACE2 binding affinity [55]. It has been noted that certain mutations in the new variants affect genomic targets used by the PCR tests. This may affect the ability of some tests to detect the virus. However, because most PCR tests detect more than one gene target, there is a small chance of a false negative result. Further, the mutations in various viral genes that help build glycan chains may influence the individual immune response, which is a serious concern relating to the antibody response to vaccines.
Recently, the actual sequence of the Pfizer mRNA vaccine was released [56]. The main difference in the vaccine code is that uracil has been replaced by 1-methyl-3’-pseudouridylyl, labelled as Ψ [57]. As such, the Ψ is accepted as normal uracil by the immune systems for the translation, transcription, and replication reactions related to the cell functions. But the substitutions stabilize the S structure, and the deployment of special 5’ and 3’ untranslated regions before and after the main spike sequence. Whereas the Oxford-AstraZeneca vaccine contains the genetic material of the SARS-CoV-2 virus S protein. The new SARS-CoV-2 variants do not appear to affect the S protein significantly to prevent immune response to the COVID-19 vaccines.
Immune Escape Variants and Disease Severity
Several genetic variants associated with immune escape have emerged in global populations. An immune escape variant of the SARS-CoV-2 is a mutation in the virus that allows it to evade the immune system. The CSIR Institute of Genomics and Integrative Biology (CSIR-IGIB) team has identified 120 ‘immune escape variants’ in SARS-CoV-2 from across the globe [58]. In addition, the team has identified at least 19 genetic variants including the S:N440K variant, which have ability to evade neutralising antibodies. Further, the team found 24 immune escape associated variants present in almost 70 per cent of the viruses sequenced from Australia. The analysis also suggests that N501Y mutation was present in a total of 290 genomes, including genomes from Australia, South Africa, USA, Denmark, and Brazil. It has been conjectured that the high rate of mutations in a virus may bring about its extinction. The speculation has been applied by Banerjee et al to SARS-CoV-2 with the hypothesis that the viral S glycoprotein is the key to receptor binding, fusion, and cell entry leading to host cell infection, and increasing spike mutations may lower the viral fitness [59]. The authors used the genomic sequences of 630 Indian isolates retrieved from the GISAID database and found 41 and 22 mutations in the S1 and S2 subunits of the spike, respectively. Tracing the mutations showed that over two-thirds had lower spike-receptor stability than the ancestral genome, which implied that the rapid build-up of mutations in the recent variants leads to a loss of fitness compared to their ancestors. Thus, irreversible accumulation of harmful mutations causes a species to die out gradually. Similarly, accumulation of beneficial mutations can lead to enhanced viral fitness.
The number of infected cases, deaths, and mortality rates related to COVID-19 vary from country to country. The reasons of these differences are several [60]. In a study, Toyoshima et al identified a total of 1234 mutations by comparing with the reference SARS-CoV-2 sequence. Through a hierarchical clustering based on the mutant frequencies, the 28 countries were classified into three clusters showing different fatality rates of COVID-19. In the analysis, the authors correlated the ORF1ab 4715L and S protein 614G variants with fatality rates. They documented that BCG-vaccination status was significantly associated with the fatality rates as well as number of infected cases in a community. In addition, the frequency of certain HLA alleles, including HLA-A*11:01, was significantly associated with the fatality rates. These findings suggest that apart from SARS-CoV-2 mutations, BCG-vaccination status and a host genetic factors including HLA genotypes might affect the susceptibility to SARS-CoV-2 infection or severity of COVID-19.
The Impact of SARS-CoV-2 Variants
SARS-CoV-2, the virus that causes COVID-19, has had a major impact on human health globally by infecting large number of people, causing severe disease in some, and being associated health sequelae resulting in short-term as well as long-term morbidity and excess mortality, especially among older and vulnerable populations. It has led to interruption of routine healthcare services, trade, education and many other societal functions, and travel disruptions. Multiple SARS-CoV-2 variants are circulating globally, and several new variants have emerged lately in the last quarter of 2020.
Some of the potential consequences of emerging variants are the following:
a) Rapid transmission - There is already evidence that one mutation, D614G, confers increased ability to spread more quickly than the wild-type SARS-CoV-2. Other variants, the British VUI-202012/01 variant and the South African 5O1Y.V2 Variant are also associated with enhanced infectivity.
b) Disease severity - There is no evidence so far that the recently identified SARS-CoV-2 variants cause more severe disease than earlier ones.
c) Ability to evade detection – The variants may evade certain diagnostic tests. However, most commercial PCR tests have multiple targets to detect the virus, so if the mutations impact some targets, the PCR will still work.
d) Likelihood of decreased susceptibility to antiviral drugs such as remdesivir and favipiravir, and other therapeutic agents such as monoclonal antibodies.
e) Likelihood of evading natural or vaccine-induced immunity. The multiple mutations in the S protein may confer ability to evade immunity induced by vaccines or by natural infection.
The Impact of SARS-CoV-2 Variants
The WHO is working with various countries to identify how current surveillance systems can be amplified and adapted to evaluate potential virus variations through ongoing systematic clinical and epidemiologic surveillance. In addition, it aims to establish of genetic sequencing capacity where possible and providing access to analyze samples for genomic sequencing and phylogenetic analysis. As part of WHO’s SARS-CoV-2 global laboratory network, the Virus Evolution Working Group (VEWG), established in June 2020, is a specific operating group for the virus evolution related observation and analysis.
The VEWG aims to –
a) Strengthen mechanisms to identify and prioritize potentially relevant mutations.
b) Early detection of relevant mutations and their potential impacts related to viral transmission and virulence, with relevance to available and prospective countermeasures including diagnostics and therapeutics.
c) Study of the impact of specific mutations, including in vitro and in vivo studies of variants in laboratory.
d) Sharing of full genome sequences and facilitating detailed analyses by scientists with expertise.
Simultaneously, there is need to reduce transmission of SARSCoV- 2 by using established disease control measures as well as avoiding introductions to animal populations as part of the global strategy to reduce the occurrence of mutations that may have potential negative public health implications [61]. Further, the increased transmissibility has potential for higher case incidence, leading to increased COVID-19 related hospitalizations, morbidity, and deaths. As warned by the WHO, a more intensive public health response may be necessary to control variant transmission.
Footnotes
a) Dr Vinod Nikhra - Affiliation – Senior Chief Medical Officer and Consultant, Department of Medicine, Hindu Rao Hospital and NDMC Medical College, New Delhi, India
b) Email: drvinodnikhra@gmail.com Alt. email: drvinodnikhra@rediffmail.com
c) ORCiD: orcid.org/0000-0003-0859-5232
d) Disclosures - None
e) The Table 1 and 2 in this Review Paper are subject to Copyright by Dr Vinod Nikhra.
References
- (2020) Max Planck Institute for the Science of Human History. COVID-19 is here to stay for the foreseeable future: Future of field-based sciences in the time of coronavirus. ScienceDaily.
- Nikhra V (2021) Exploring COVID-19: Relating the Spike Protein to Infectivity, Pathogenicity, and Immunogenicity. ResearchGate.
- Van Regenmortel MH, Mahy BW (2004) Emerging issues in virus taxonomy. Emerg Infect Dis 10(1): 8-13.
- Walker PJ, Siddell SG, Lefkowitz EJ (2020) Changes to virus taxonomy and the Statutes ratified by the International Committee on Taxonomy of Viruses (2020). Arch Virol. 165(11): 2737-2748.
- Kuhn JH, Bao Y, Sina Bavari S (2012) Virus nomenclature below the species level: a standardized nomenclature for natural variants of viruses assigned to the family Filoviridae. Archives of Virology 158(1): 301-311.
- https://www.gisaid.org/references/statements-clarifications/clade-and-lineage-nomenclature-aids-in-genomicepidemiology-of-active-hcov-19-viruses/
- https://nextstrain.org/sars-cov-2/
- Guan Q, Sadykova M, Mfarreja S (2020) Genetic barcode of SARS-CoV-2 for monitoring global distribution of different clades during the COVID-19 pandemic. International Journal of Infectious Diseases 100: 216-223.
- Rambaut A, Loman N, Pybus O (2020) On behalf of COVID-19 Genomics Consortium UK (CoG-UK). Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations. SARS-CoV-2 coronavirus, nCoV-2019 Genomic Epidemiology.
- Koyama T, Platt D, Parida L (2020) Variant analysis of SARS-CoV-2 genomes. Bulletin of the World Health Organization 98(7): 495-504.
- Naqvi AAT, Fatima K, Mohammad T (2020) Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1866(10): 165878.
- Robson F, Khan KS, Le TK (2020) Coronavirus RNA Proofreading: Molecular Basis and Therapeutic Targeting. Molecular Cell 79(5): 710-727.
- Andrés C, Garcia Cehic D, Gregori J (2020) Naturally occurring SARS-CoV-2 gene deletions close to the spike S1/S2 cleavage site in the viral quasi species of COVID19 patients. Emerg Microbes Infect 9(1): 1900-1911.
- SA Kemp, RP Datir, DA Collier (2020) Recurrent emergence and transmission of a SARS-CoV-2 Spike deletion ΔH69/ΔV70. bioRxiv preprint.
- Wang F, Huang S, Gao R (2020) Initial whole-genome sequencing and analysis of the host genetic contribution to COVID-19 severity and susceptibility. Cell Discov 6(1): 83.
- Yang HC, Chen CV, Wang JH (2020) Analysis of genomic distributions of SARS-CoV-2 reveals a dominant strain type with strong allelic associations. PNAS 117(48): 30679-30686.
- Baric RS (2020) Emergence of a Highly Fit SARS-CoV-2 Variant. NEJM. 383(27): 2684-2686.
- Korber B, Fischer WM, Gnanakaran S (2020) Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 Virus. Cell 182(4): 812-827.
- Zhang L, Jackson CB, Mou H (2020) The D614G mutation in the SARS-CoV-2 spike protein reduces S1 shedding and increases infectivity. bioRxiv preprint.
- Plante JA, Liu Y, Liu J (2020) Spike mutation D614G alters SARS-CoV-2 fitness and neutralization susceptibility. Nature.
- Hou YJ, Chiba S, Halfmann P (2020) SARS-CoV-2 D614G Variant Exhibits Enhanced Replication ex vivo and Earlier Transmission in vivo. Preprint. bioRxiv 9(28): 317685.
- Zhang L, Jackson CB, Mou H (2020) SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat Commun 11: 6013.
- (2020) Threat Assessment Brief: Rapid increase of a SARS-CoV-2 variant with multiple spike protein mutations observed in the United Kingdom (PDF) (Report). European Centre for Disease Prevention and Control (ECDC).
- (2020) Emerging SARS-CoV-2 Variants in UK Like the B.1.1.7 Strain and the 501.V2 Strain in South Africa And Elsewhere Will Change Course Of COVID-19 Badly.
- European CDC on B.1.1.7 mutant.
- Chand M, Hopkins S, Dabrera G (2020) Investigation of novel SARS-COV-2 variant: Variant of Concern 202012/01 (Report). Public Health England.
- CDC on B.1.1.7 mutant.
- (2020) COG-UK update on SARS-CoV-2 Spike mutations of special interest | Report 1 (PDF) (Report). COVID-19 Genomics UK Consortium (COG-UK). pp. 7.
- https://www.sciencemediacentre.org/expert-reaction-to-pms-and-cmos-statements-about-new-restrictions-and-the-new-sars-cov-2-variant/
- WHO - SARS-CoV-2 Variants.
- Greaney AJ, Loes AN, Crawford KHD (2020) Comprehensive mapping of mutations to the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human serum antibodies. biorxiv preprint.
- WebMD Health News. New COVID ‘Super Strains’ Could Disrupt Life Again.
- (2020) WHO - SARS-CoV-2 mink-associated variant strain – Denmark, Disease Outbreak News.
- (2020) Detection of new SARS-CoV-2 variants related to mink.
- (2020) Nigeria identifies new variant of COVID-19 in samples collected in August: Report. Devdiscourse News Desk Updated 22-12-2020.
- Pereira F (2020) Evolutionary dynamics of the SARS-CoV-2 ORF8 accessory gene. Infect Genet Evol 85: 104525.
- Jolly B, Rophina M, Shamnath A (2020) Genetic epidemiology of variants associated with immune escape from global SARS-CoV-2 genomes. bioRxiv preprint.
- Yi C, Sun X, Ye J (2020) Key residues of the receptor binding motif in the spike protein of SARS-CoV-2 that interact with ACE2 and neutralizing antibodies. Cellular & Molecular Immunology 17(6): 621-630.
- Ou J, Zhou Z, Dai R, Zhang J, Lan W, et al. (2020) Emergence of RBD mutations in circulating SARS-CoV-2 strains enhancing the structural stability and human ACE2 receptor affinity of the spike protein. [preprint]. Cold Spring Habor: medRxiv
- Koyama T, Weeraratne D, Snowdon JL, Parida L (2020) Emergence of drift variants that may affect COVID-19 vaccine development and antibody treatment. Pathogens 9(5): 324.
- Mercatelli D, Giorgi FM (2020) Geographic and Genomic Distribution of SARS-CoV-2 Mutations. Front. Microbiol 11: 1800.
- Day T, Gandon S, Lion S (2020) On the evolutionary epidemiology of SARS-CoV-2. Current Biology 30: R849-R857.
- (2020) Africa Centres for Disease Control and Prevention, Latest updates on the COVID-19 crisis in Africa.
- Walker PG, Whittaker C, Watson O (2020) Report 12 - The global impact of COVID-19 and strategies for mitigation and suppression.
- Uyoga S, Adetifa IMO, Karanja HK (2021) Seroprevalence of anti-SARS-CoV-2 IgG antibodies in Kenyan blood donors. Science 371(6524): 79-82.
- Frey A (2020) Mozambique: 3.79 per cent of Maputo residents exposed to coronavirus. Club of Mozambique.
- https://www.businesstoday.in/latest/trends/india-may-have-missed-90-infections-for-every-detected-covid-19-case/story/425175.html
- Florindo HF, Kleiner R, Vaskovich Koubi D (2020) Immune-mediated approaches against COVID-19. Nat. Nanotechnol 15(8): 630-645.
- Poland GA, Ovsyannikova IG, Kennedy RB (2020) SARS-CoV-2 immunity: review and applications to phase 3 vaccine candidates. Lancet 396(10262): 1595-1606.
- Tillett RL, Sevinsky JR, Hartley PD (2020) Genomic evidence for reinfection with SARS-CoV-2: a case study. The Lancet Inf Dis 21(1): 52-58.
- (2021) Health and Science News. The UK is delaying second vaccine shots and it’s proving controversial. Here’s what we know so far.
- (2021) Opinions - It’s time to consider delaying the second dose of coronavirus vaccine.
- (2021) WHO: 2nd dose of BioNTech/Pfizer vaccine can be delayed. It can be given up to six weeks after the first, the health body says.
- Shang J, Ye G, Shi K (2020) Structural basis of receptor recognition by SARS-CoV-2. Nature 581(7807): 221-224.
- Qiao B, De la Cruz MO (2020) Enhanced Binding of SARS-CoV-2 Spike Protein to Receptor by Distal Polybasic Cleavage Sites. ACS Nano 14(8): 10616-10623.
- (2020) Information for Healthcare Professionals on Pfizer/BioNTech COVID-19 vaccine’. Medicines and Healthcare products Regulatory Agency (MHRA).
- (2020) World Health Organization. "Messenger RNA encoding the full-length SARS-CoV-2 spike glycoprotein" (DOC). WHO MedNet.
- Rophina M, Pandhare K, Sethi M, et al. (2020) Comprehensive resource of genetic variants in SARS-CoV-2 genomes from India Web Resource.
- Banerjee R, Basak K, Ghosh A, et al. Spike Protein Mutational Landscape in India: Could Mullers Ratchet Be A Future Game-Changer For COVID-19? bioRxiv preprint.
- Toyoshima Y, Nemoto K, Matsumoto S (2020) SARS-CoV-2 genomic variations associated with mortality rate of COVID-19. Journal of Human Genetics 65: 1075-1082.
- Kunzmann K (2021) WHO Shares Update, Guidance on New SARS-CoV-2 Variants.
