 Case Report
Case ReportAbstract
Objective: The main objective of our article is to present the case of a pheochromocytoma as a rareneuroendocrine tumour which can be sporadic or hereditary form.
Case: We present a case of a 34-year-old woman admitted at endocrinology department forexploration of a secondary resistant hypertension and where imaging had shown a giant adrenalmass attributed to an invasive pheochromocytoma. Otherwise, morph typic exam of the patientshowed features of a NOONAN syndrome associated with a neurofibromatosis type 1.
Conclusion: each case is a particular case which requires special attention and a careful clinicalexamination to be able to reveal its mystery.
Keywords: Adrenal Medulla; Cafeau- Lait Spots; Pheochromocytoma; Neurofibromatosis 1;Noonan Syndrome
Introduction
Pheochromocytoma is a relatively rare neuroendocrine tumour which can be sporadic or fitting into apolymorphic syndromic context. These syndromes include: Multiple endocrine neoplasia, Von-HippelLindau disease, and neurofibromatosis type 1 (NF1)[1]. We report a case of a pheochromocytoma in a young woman in the context of neurofibromatosistype 1 associated with a rare genetic syndrome: the NOONAN syndrome (NS).NF1 is the most common type of neurofibromatosis, accounting for about 90% of all the cases. It isa frequent autosomal dominant genetic disease with various clinical manifestations and a weakgenotype-phenotype correlation. The diagnosis of NF1 is based on clinical findings with reference tospecific criteria.Genetic testing is not recommended for diagnosis [2,3].On the other hand, NS is a heterogeneous relatively frequent genetic disorder, with an estimatedprevalence between 1/1000 and 1/2500 live births. It is transmitted in an autosomal dominant waywith a variable phenotypic expression [4].The particularity of the case presented here is about the coexistence of NF1 and NS: an associationwhose mechanisms are not yet wellunderstood.
Observation
A 34-year-old female, with no personal or family history, was examined by a primary-care physicianfor having a gravida hypertension that has persisted6 months after her delivery. Despite thecombination of 3 antihypertensive drugs, blood pressure was still poorly controlled. Accordingly, thepatient was admitted to our department for a diagnosis of a secondary hypertension.The patient complained of an holocranial headache with intermittent attacks of palpitations and aprofuse sweating since the last month. She also reported having an hypoacusis and a recurrentepistaxis. The physical examination showed that her blood pressure was 210/110mmHg controlled at 140/90mmHg after taking her antihypertensive treatment without complaining of a dyspnoea or a chest pain.She was also found to have tachycardia at 111bpm.The electrocardiogram test uncovered a left ventricular hypertrophy with an elevated Sokolow-Lyonindex calculated at 40mm, without arrythmia.Morphologically, the patient had a short stature (Height: 154cm; Weight: 54kg), a scoliosis, a chestdeformation with widely spaced and low set nipples, an hypertelorism, low-set ears, and a mildmental retardation. As skin manifestations, there was multiple « café-au-lait » spots sized over 1.5cm which have progressed since the childhood. In addition to these spots, there were unesthetic and pruriginous subcutaneous and cutaneous neurofibromas on the trunk and the arms anddisseminatedfreckling’s with accentuation in skinfold areas (armpit and groin) (Figures 1&2) The eye examination indicated that the patient had a Stage IV hypertensive retinopathy with thepresence of specific Lisch nodules.
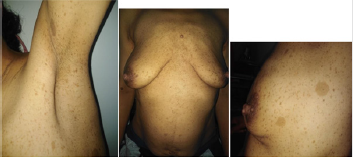
Figure 1: Multiple café-au-lait spots and disseminatedfreckling’s.
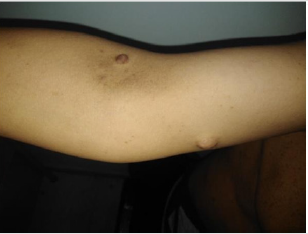
Figure 2: Cutaneous neurofibromas.
The ENT examination legitimized by the hypoacusis showed a retro tympanic air-fluid level and abilateral presbycusis on audiometry.Otherwise, given our patient’s young age, morphotype, and evident skin lesions, we suspected agenetic syndrome, especially since her baby who is a 6 month-old girl presents a typical facialdysmorphism of a genetic syndrome, multiple « café au lait » spots, and a cutaneous angioma.All these clinical findings raise the suspicion of neurofibromatosis type 1 associated with NS.The biochemistry results pinpointed that the patient had a prediabetes status (Fasting plasmaglycemia = 1,05g/l: Glycated hemoglobin=6,2%), a normal renal function, no hypokalemia, and anormocytic normochromic anemia.The three days-measurement of urinary fractionated metanephrons revealed elevated levels ofnormetanephrines (greater than 22 times the upper limit of normal) and metanephrons (up to 130times the upper reference value) (Figure 3).An abdominal ultrasound unfolded a welldefined round adrenal right nodule of heterogeneoushyperechoic echostructure, vascularized (with color-doppler), and sized 73x70mm. These signs aresuggestive of a pheochromocytoma (Figure 4).
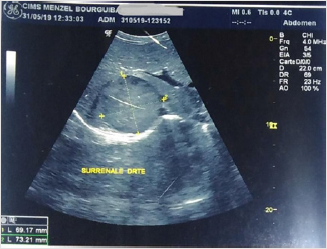
Figure 3: Ultrasound section of a 7cm right adrenal mass.
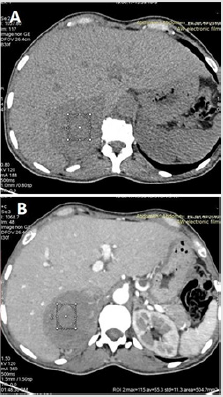
Figure 4: A CT scan showing the right adrenal pheochromocytoma without (A) and with contrast (B).
A CT scan displayed a well-defined rounded adrenal mass, spontaneously hypodense, enhanced atarterial time, and with necrosis areas measuring 73 x 70 mm and extending over 70 mm in height. The mass meets the inferior vena cava and the right renal vein which arecompressed and pushed back forward with loss of fatty edging. It is associated with perirenalcollaterals (Figure 5) A MIBG scanshowed an intense and heterogeneous uptake at the level of the right adrenal mass 112described on the CT scan without a remote fixation on the contralateral adrenal gland (Figure 6).Our patient was treated with verapamil, monoxide, alpha-blocker and then with an association ofa beta-blocker as a preparation for the surgery.After two weeks of preparation, a safety sub-costal adrenalectomy was performed. The pathologicalexamination of the mass showed histo-morphological features compatible with a possibly aggressivepheochromocytoma with a PASS score=4. It also indicated the presence of capsular and vascularinvasion (Figures 7&8).After surgery, the patient was normotensive without any treatment. Her urinary fractionedmetanephron level returned to normal.A genetic testing was advised for our patient and her baby for an early diagnosis to providean adequate follow-up. However, this test is not available in our country
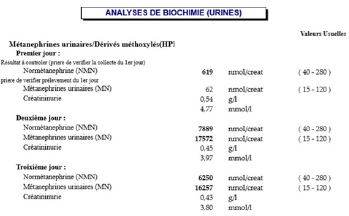
Figure 5: Rate of urinary fractioned metanephrines during 3 days in our patient.

Figure 6: MIBG scan showing an intense uptake of I-123 meta-iodobenzylguanidine in the rightadrenal mass.
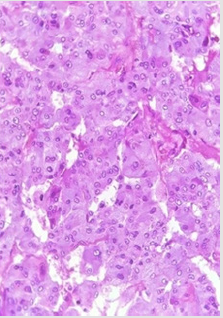
Figure 7: Pheochromocytoma, showing the nested arrangement of cells (Zellballen) and stippledchromatin. H&E stain X 400.
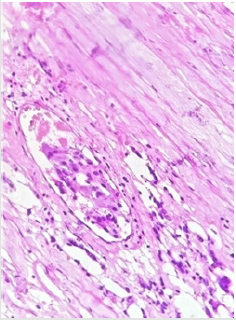
Figure 8: Tumor embolism in a pericapsular vessel. H&E stain X 400.
Discussion
Our observation illustrates the case of a 34-year-old patient complaining of a persistent gravidahypertension, resistant to a triple therapy and having visceral impact made of hypertensiveretinopathystageIV andanelectricleft ventricularhypertrophy.Anon-metastaticpheochromocytoma included in the context of type 1 neurofibromatosis associated with a NS isaccounting for this case.** Pheochromocytoma is a rare neuroendocrine tumor arising from chromaffin cells of the adrenalmedulla and characterized by a hypersecretion of catecholamines. The incidence is approximately 2-8 per 1 million adults-years. The higher frequency is noticed during the fourth decade [5].In general, Pheochromocytomamay be a sporadic tumor or a part of ageneticsyndrome.Hereditary forms are about 30-40% of the cases with a prevalence of 1 case per 500,000 adults [5].Up to now, the genetics of pheochromocytoma have been divided into 3 main groups:Multipleendocrine neoplasia type II, Von Hippel-Lindau disease, and Neurofibromatosis type 1 wherepheochromocytoma is not firstly seen [1].
Currently, family history is the most important predictor factor of the existence of a germlinemutation in individuals with pheochromocytoma, about 90% of whom have a family history with anidentifiable mutation. However, the absence of a family history should not preclude the indication ofa genetic assessment[5].Our patient, young, without family history, has a pheochromocytoma associated with a specialmorphotype and skin lesions which lead to the suspicion of a NF1 associated with a NS.The diagnosis of a hereditary pheochromocytoma syndrome should be suspected in any individualwith a diagnosis of pheochromocytoma. In clinical practice, patients with pheochromocytoma canshow particularities leading to think about a hereditary form. Such particularities include a positivefamily history (based on the family pedigree or on the identification of a pheochromocytomasusceptibility gene mutation in a relative), syndromic features, and multifocal, bilateral, Recurrent,metastatic disease, Extra-adrenal, or early onset (age <45 years). (The most known familialautosomal dominant diseases which can be clinically suspected are: Neurofibromatosis type 1 (NF1),multiple endocrine neoplasia type 2 (MEN2), von Hippel-Lindau(VHL) syndrome, and renal cellcarcinoma with SDHB mutation [6,7].
According to the 1st publication by Neumann et al. genotyping of the principal implicated genes inpheochromocytoma (SDHB, SDHD, VHL, RET) has been performed in eight studies, each comprisingover 200 patients and including 3694 subjects with 1250 germinal mutations (33.8%). This increasedfrequency defends the indication of genetic testing in all cases of pheochromocytoma. The most164common mutations are for: SDHB (10.3%), SDHD (8.9%), VHL (7.3%), RET (6.3%), and NF1 (3.3%)[7].**NF1, also known as Von Recklinghausen’s disease, is the most common type of neurofibromatosisaccounting for about 90% of all the cases. It is a frequent human genetic disease, with a prevalenceranging from 1/3000 to 1/4000 births. It is inherited as an autosomal dominant trait with a completepenetrance and a variable phenotypic expression [2].It affects many systems under the name of neurocutaneous syndrome. These are: The neurological,the cardiovascular, the gastrointestinal, the endocrine, and the orthopedic systems. The NF1 resultsfrom a point mutation or a deletion of the NF1 gene located on chromosome 17q11.2. The geneimplicated is a tumor suppressor gene via its product: Neurofibromin. About 50% of the cases arehereditary and 50% of them are de novo mutations [3,8,9].The clinical manifestations are diverse with the association of skin signs (« café au lait » spots in 95%of the cases, frecklings) and Lisch nodules in the iris and cutaneous and/or subcutaneousneurofibromas (40 - 60% of the cases) [3].
NF1 has a weak genotype-phenotype correlation and a high inter and intra-familial phenotypicvariability [10].The diagnosis of NF1 is based on clinical findings with reference to criteria developed by “the UnitedStates National Institutes of Health (NIH)” which were published in 1987 and have remained valid. Apatient must meet at least 2 of the following criteria to make the diagnosis of NF1(Table 1)[2,3].The diagnosis of patients with neurofibromatosis is based on clinical findings and genetic tests aregenerally unnecessary [3,5,11].The diagnosis of NF1 is frequently delayed and can be made when meeting other signs. This appliesto the case of our patient who fulfilled 5 of the 7 diagnostic criteria for NF1 ( six café-au-lait spots, sixdermal neurofibromas, axillary and inguinal frecklings, Lisch nodules, and a suspected first-degreerelative who is her child).Therefore the diagnosis was only suspected after the confirmation ofpheochromocytoma despite the pre-existing skin lesions. Thus, the diagnosis currently seems almostobvious.The association between neurofibromatosis and pheochromocytoma was first reported by Suzuki etal. in 1910. In fact, pheochromocytoma rarely occurs during NF1 with a frequency of 0.1% to 5.7%.Such incidence seems to be higher compared to the general population. The incidence increases upto 20% in hypertensive patients [3].

Table 1: 1987 NIH Consensus Development Conference diagnostic criteria for NF1 [10].
The middle age of onset is around the fourth decade, but it can be in childhood. These tumorsprogress the same way as sporadic pheochromocytomas [5].Most pheochromocytomas during NF1 mainly produce normetanephrine and are most oftenmanifested by hypertension and noradrenergic signs. However, 22% of them remain asymptomatic200[3,5].Although rare, NF1 can be responsible for an uncontrolled hypertension with large variations as it isthe case of our patient. Morbidity will be significant with a high risk of mortality if the diagnosis isdelayed or missed .This can be due to a catecholamine release by pheochromocytoma which isresponsible for the variability and the severity of hypertension as well as the myocardial infarctionand the cardiac arrhythmias [2,3]. The diagnosis of pheochromocytoma is missed in almost 20% of the cases of NF 1 when the patient isasymptomatic. However, in case that it is associated with hypertension, the diagnosis is made in 20to 56% of the patients. The following table regroups all the publications concerning this topic andhighlights the low incidence of pheochromocytomas among the confirmed cases of NF1 (Table 2)210[2,3].Although NF1 is among the well-described genetic syndromes,including pheochromocytoma, thereare no recommendations on screening for pheochromocytomas (or paragangliomas) in knownpatients having NF1 [2,5,12].
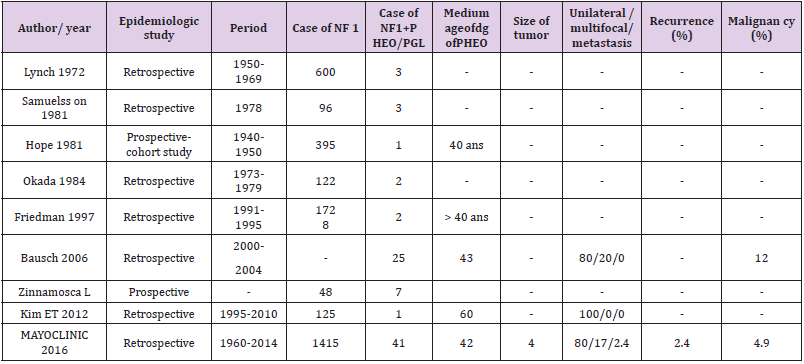
Table 2: Summary of the existing literature on the association between NF1 and PHEO / PGL [2].
As it is a complicated process, detection of mutations in the NF1 gene is available in specializedlaboratories, and the diagnosis of NF1 can be established by clinical findings alone. Nevertheless,some patients with an apparently sporadic pheochromocytoma had the NF1 mutation, all with mildmanifestations of the disease. These results show the importance of careful clinical examination ofpossible clinical signs of an underlying mutation in all the cases of pheochromocytoma [7] (Table 3).Pheochromocytoma in NF1 is unilateral and not metastatic in more than 80% of cases (and has amalignant rate of about 10%)However, the particularity presented by our case, is that the tumor, clinically and radiologicallybenign, presents histological features of malignancy with a score 4 of PASS, vascular embolisms, anda focal capsular invasion.According to many authors, a PASS score <4 is more in favor of benign tumors and a PASS score ≥ 4 israther in favor of potentially malignant tumors. Others assume that tumors with a PASS score ≥ 4should be closely followed for a possible recurrence and those with a PASS score ≥ 6 are potentiallymalignant [2,5,12].
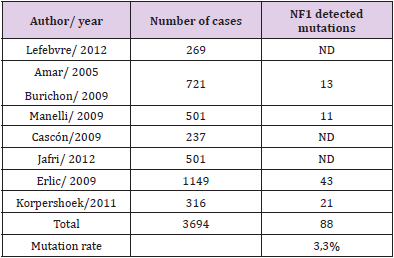
Table 3: Summarized.
However, this prognostic score, which is the most used, is not yet validated and is not recommendedin current clinical practice, because it does not allow a clear histological diagnosis of benign andmalignant tumors, as well as the importance of inter and intra-anatomopathological variability235[12,13].The combination of the facial dysmorphism is seen in our patient with a small size, a chestdeformation associated with skin lesions, that is why a NS was suspected.**NS is a heterogeneous genetic disorder, relatively frequent with an estimated prevalence between1/1000 and 1/2500 live births. It occurs sporadically in 60% of the cases or as mendelian traittransmitted in an autosomal dominant way with a complete penetrance and a variable phenotypicexpression [4].This syndrome is caused by mutations in the PTPN11, SOSI, KRAS, NRAS, RAF1, BRAF, and MEK1(MAP2K1) genes. These mutations are involved in the signal transduction pathway Ras/MAPK(protein kinase activated by a mitogen) and they currently explain around 70% of the affectedpersons [14].Its clinical manifestations include a typical dysmorphic facial which evolves with age, webbing of theneck, postnatal reduced growth, chest deformation, congenital heart disease (present in 65 to 85% ofpatients with NF1: pulmonary valve stenosis (50-65%), cardiomyopathy hypertrophic (20%), inter-atrial communication (10-25%), inter-ventricular communication (5%), persistent arterial duct (3%)),eye abnormalities, cryptorchidism, and skeletal anomalies associated with other comorbidities [15].This phenotype becomes less evident with age causing the diagnosis sometimes to be difficult inadulthood [16].NS often remains a clinical diagnosis. Due to the variability of its clinical presentation and theheterogeneity of the manifestations, it is important to provide a multidisciplinary care [16].
Our patient had a low IQ without other neurological manifestations. The audiometry showedbilateral presbycusis and retro-tympanic bubbles which signal a retro tympanic effusion (Acute otitismedia or sequelae) and a scoliosis on skeletal examination. Her skeleton x-ray, done in search ofbone dysplasia or cortical improvement, showed an osteoarticular integrity, an absence of corticalimprovement and an osteocondensing lesion of the upper epiphyses of the tibia described as abenign condensing island. The patient also had a history of epistaxis without other hemorrhagicsymptoms and no cardiac, endocrine, gastrointestinal, or genitourinary related disorders. In addition,she had no manifestation of a malignant hyperthermia during the surgery under general anesthesia.Various theories have been proposed to explain the coexistence of NF1 and NS. Because each ofthese disorders is common in the general population, it is possible that the two could occur together.Alternatively, the neurofibromatosis- NS combination may be an entity. Some previousreports have described patients with neurofibromatosis 1 with the Noonan’s phenotype [15,17].The coexistence of NF1 and NS was explained by a coincidence of two different autosomal disorders.Some manifestations of NF1 (café-au-lait spots) can occur as a symptom of a classical NS. Themanifestations of NS in these patients are a variable form of NF1 or a discrete form of NFNS [18].The NFNS phenotype is highly heterogeneous at both the clinical and the molecular levels. Manyother causes may be intricate in the occurrence of neurofibromatosis 1 and NS [17].Our case was unusual in that the patient was presented with pheochromocytoma at the age of 34years and had not been diagnosed with neurofibromatosis– NS before.However, the main problem remains with her little baby in whom the NS was suspected. Hence theimportance of the multidisciplinary care, specializing in pediatrics and genetics, in order not tomisdiagnose associated comorbidities.
Conclusion
Although rare in patients with NF1, pheochromocytoma can generate a poorly controlledhypertension with the risk of developing other cardiovascular complications that should beinvestigated in time. Whether sporadic or involved in type 1 neurofibromatosis, as in the case of ourpatient, its management remains the same.The presentation of our case underlines the importance of careful examination for the clinicalfeatures of neurofibromatosis and the phenotypic traits of associated diseases in patients withpheochromocytoma.
Declaration of Interest
There is no conflict of interest.
Funding
This research did not receive any specific grant from any funding agency in the public,commercial or not-for-profit sector.
Author Contribution Statement
Pr.JRAD who contributed to the selection of CT-scansPr. Rammeh and her team selected histological sections and gave the comments below. The article was written in collaboration with Dr.ROJBI, edited by members of the endocrinologyteam.
Acknowledgements
A special acknowledgment to all contributors.
References
- Gimenez Roqueplo (2003) Paragangliomes et phéochromocytomes 64(5): 396‑
- Gruber LM, Erickson D, Babovic-Vuksanovic D, Thompson GB, Young WF, et al. (2017) Pheochromocytoma and paraganglioma in patients with neurofibromatosis type 1. J Clin Endocrinol Metab. janv 86(1): 141-149.
- Petrovska J, Gerasimovska Kitanovska B, Bogdanovska S, Pavleska Kuzmanoska S (2015) Pheochromocytoma and Neurofibromatosis Type 1 in a Patient with Hypertension. Open Access Maced J Med Sci. 3(1): 713.
- Piard J (2009) Syndrome de Noonan et apparentés: Etude clinique et moléculaire de 51 enfants etadultes pris en charge au centre de référence « “anomalies de développement” » de Nancy. [NANCY]: UNIVERSITE HENRI-Pü
- Geurts JL, Rich TA, Evans DB, Wang TS (2016) Hereditary Syndromes Involving Pheochromocytoma and Paraganglioma. In: Genetic Diagnosis of Endocrine Disorders [Internet]. Elsevier pp. 221‑2
- Else T, Greenberg S, Fishbein L (2008) Hereditary Paraganglioma-Pheochromocytoma Syndromes. Gene Reviews pp. 32.
- Lenders JWM, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SKG, et al. (2014) Pheochromocytoma and Paraganglioma: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab 99(6): 1915‑19
- Yoorim Seo, Yeonjeong Jeong, Dong Yoon Kim, Kyueun Choi, Eun Sook Kim, et al. (2015) A novel neurofibromatosis type 1 (NF1) mutation in a patient with NF1 and pheochromocytoma. Korean J Intern Med 33(1): 214‑21
- Remón-Ruiz P, Aliaga-Verdugo A, Guerrero-Vázquez R (2017) Pheochromocytoma in neurofibromatosis type 1 during pregnancy. Gynecol Endocrinol. févr 33(2): 93‑9
- Gutmann DH, Ferner RE, Listernick RH, Korf BR, Wolters PL, et al. (2017) Neurofibromatosis type 1. Nat Rev Dis Primer 3(1): 17004.
- Anderson JL, Gutmann DH (2015) Neurofibromatosis type 1. In: Handbook of Clinical Neurology [Internet]. Elsevier 132: 75‑
- Mlika M, Kourda N, Zorgati MM, Bahri S, Ammar SB, et al. (2013) La valeur pronostique du PASS score dans la distinction entre phéochromocytome bénin et malin. Tunis Med 91(3): 209‑2
- Savoie PH, Murez T, Fléchon A, Sèbe P, Rocher L, et al. (2018) RETRACTED: Recommandations françaises du Comité de Cancérologie de l’AFU - Actualisation 2018-2020: tumeur de la surré Prog En Urol 28(12): S175‑S193.
- H Chen (2017) Noonan syndrome. Medline Plus pp. 2151‑21
- Tartaglia M, Zampino G, Gelb BD (2010) Noonan Syndrome: Clinical Aspects and Molecular Pathogenesis. Mol Syndromol 1(1): 2‑
- Romano AA, Allanson JE, Dahlgren J, Gelb BD, Hall B, et al. (2010) Noonan Syndrome: Clinical Features, Diagnosis, and Management Guidelines. Am Acad Pediatr 126(4): 746‑7
- Agras PI, Baskin E, Sakallioglu AE, Arda IS, Ayter S, et al. (2003) Neurofibromatosis-Noonan’s Syndrome With Associated Rhabdomyosarcoma of the Urinary Bladder in an Infant: Case Report. J Child Neurol. janv 18(1): 68‑
- Yazdizadeh M, Tapia JL, Baharvand M, Radfar L (2004) A case of neurofibromatosis-Noonan syndrome with a central giant cell granuloma. Oral Surg Oral Med Oral Pathol Oral Radiol Endodontology 98(3): 316‑320.
