 Case Report
Case ReportAbstract
Joubert Syndrome and related disorders encompass a spectrum of rare autosomal recessive diseases which have “Molar Toth” appearance of midbrain and clefted hypoplastic vermis as obligatory imaging markers. They may be associated with various “Oculo-faciodigital” and visceral defects. We present a similar case of a 1-year old boy who presented with global developmental delay and was found to have “Joubert Syndrome” on MRI brain. Lack of any extracranial features pointed towards “Classical” or “Pure Joubert Syndrome”. The parents were explained the nature of the disease and were advised regular follow up. The case report highlights the classical imaging features which form the basis of optimal diagnosis of this rare disease.
Keywords: Molar Tooth; Joubert; Clefted Vermis
Introduction
Joubert syndrome is a rare disorder characterized by the classical “Molar Tooth” appearance of Midbrain on axial sections of brain [1,2]. It is a part of spectrum of diseases known as Joubert Syndrome and Related Disorders (JSRD). Six phenotypes of JSRD have been described according to their associations with ocular, renal, oculo-renal, hepatic and oro-facio-digital defects. The Classic Joubert Syndrome is a “pure syndrome” with no extra cranial manifestation [3]. We report a case of 1-year old boy who presented with global development delay and was found to have typical features of “Joubert Syndrome” on MRI. The importance of imaging is well highlighted in establishing the diagnosis of this rare disease, especially where genetic analysis may not be possible.
Case Report
A 1-year-old boy presented to the Paediatrics department of our Institute with global developmental delay and extrapyramidal features. He was a term child born out of consanguineous marriage with an uneventful birth and family history. On examination, the infant was found to have nystagmus. No other ocular anomalies were present. No polydactyly or oro-facial defects were visualized. The patient was referred for MRI examination of brain in our Radiology Department which was carried out under sedation in Siemens Magnetom Aera 1.5 T MRI scanner. Axial MRI sections of brain revealed the characteristic “Molar Tooth” appearance of midbrain (Figure 1) with “Batwing Shaped” fourth ventricle. The cerebellar vermis was severely hypoplastic, dysmorphic and clefted (Figure 2). Sagittal MRI sections of brain showed small dysmorphic vermis with an elongated fourth ventricle. The roof of the fourth ventricle was convex upwards with an “enlarged rounded fastigial point”. Note was made of enlarged retro cerebellar space (Figure 3).
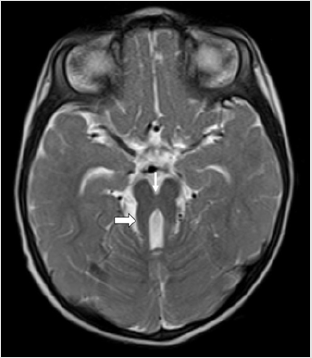
Figure 1: Axial T2 weighted (T2WI) MRI section of brain of a 1-year-old boy with global developmental delay and extrapyramidal features showing thickened superior cerebellar peduncles (thick arrow) and deep interpeduncular fossa (thin arrow) giving rise to the classical “Molar Tooth” sign of Mid brain. This sign is an obligatory imaging marker of Joubert Syndrome.
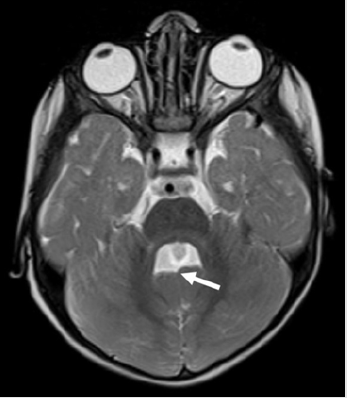
Figure 2: Axial T2WI MRI section of brain of the infant with Joubert Syndrome showing “Batwing” appearance of the fourth ventricle with small and clefted cerebellar vermis (arrow).
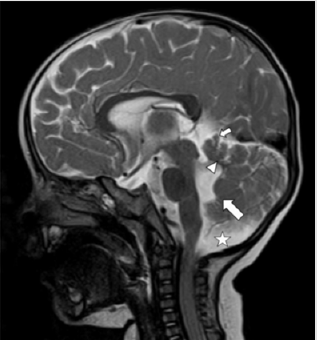
Figure 3: Mid Sagittal T2WI MRI section of brain of the infant with Joubert Syndrome showing small dysmorphic vermis (small arrow) with an elongated fourth ventricle. The roof of the fourth ventricle is convex upwards (arrowhead) with an “enlarged rounded fastigial point” (larger arrow). Note the enlarged retro cerebellar space (star).
Ultrasound abdomen done to rule out any renal and hepatic defects was unremarkable. Genetic analysis was not possible due to financial constraints of the parents. The characteristic imaging findings on a background of distinctive clinical scenario helped us to establish the diagnosis of “Joubert Syndrome”. The differential diagnosis of “Vermian hypoplasia” was ruled out due to presence of vermian cleft and other classical features of Joubert Syndrome. The cerebellar hemispheres were not fused in the midline, thus ruling out the diagnosis of “Rhombencephalosynapsis”. Due to lack of ocular abnormalities like coloboma and oro-facial-digital defects, we categorized the patient into “Classic or Pure Joubert” Syndrome. The parents were educated about the nature and prognosis of the disease, various therapies that may be needed in future and were advised to keep the child on regular follow up.
Discussion
Joubert’s Syndrome was named after a French Neurologist “Maria Joubert” who first described its findings in four siblings in 1969 [2,3]. JSRD are characterised by disturbances in cognition and facial dysmorphism. Despite the wide spectrum of neuroimaging findings described in the affected patients, vermian hypoplasia and molar tooth sign represent the “obligatory imaging biomarkers” of this entity.The marked pleomorphism in manifestations may be attributed to allelic and locus heterogeneity. JSRD predominantly include autosomal recessive disorders. Mutations in as many as 10 genes have been described in JSRD. They have been attributed to mutations of “Ciliary proteins” that affect the migration of cells [1].The clinical manifestations of Joubert syndrome and related disorders encompass intellectual impairment, developmental delay, hypotonia, ataxia, episodic alteration in respiratory rate and oculomotor apraxia. Facial dysmorphism in the form of facial hypotonia giving open mouth configuration, hypertelorism, ptosis and broadened forehead have been recorded [4]. Our patient had features of delayed development and cerebellar involvement. However, no respiratory complains or “oro-facial-digital” defects were seen.
MRI is the primary imaging modality for diagnosis of Joubert Syndrome. “Molar tooth sign” is a mandatory and classical imaging feature seen on axial sections of brain. It is attributed to the malformation of midbrain and hindbrain where elongation and thickening of the superior cerebellar peduncles is seen along with a deep inter-peduncular fossa [1–3, 5]. Absence of decussation of fibres of superior cerebellar peduncles is the cause of deep interpeduncular fossa of mid brain. Using diffusion tensor imaging, this has been well illustrated via fibre tracking by Poretti et al [6]. Functional MRI has provided complementary information by recording abnormal activation patterns of brainstem nuclei. The other significant imaging feature of the disease includes “Clefted Vermis” with a “batwing Appearance” of Fourth ventricle. Midline sagittal sections of MRI show a small vermis which is dysmorphic with a deformed forth ventricle. The fourth ventricle is elongated, shows upward convexity of its roof and an enlarged rounded fastigium. Our case showed all these classical features of Joubert Syndrome on MRI imaging.Additional infratentorial manifestations include enlargement of the posterior cranial fossa, increased retrocerebellar CSF space along with hypoplastic as well as dysplastic cerebellar hemispheres.
An increased association with supra-tentorial manifestations has been established in patients with Oral-Facial-Digital syndrome type VI including corpus callosal agenesis/dysgenesis, septum pellucidum agenesis, hypothalamic hamartomas and derangements in developmental neuronal migration [7].Management of children with JSRD requires a multi-disciplinary holistic approach equipped with parental education, financial preparedness, customised physical and speech therapy sessions and most importantly, patience for the attainment of developmental milestones. Despite the discordance in genotype-phenotype, Genetic counselling and parental education are important components in the management of this spectrum.
Conclusion
JSRD are a rare group of disorders with classical imaging markers which form the basis of optimal diagnosis. The characteristic MRI pattern of “Molar Tooth” sign with vermian hypoplasia should encourage us to rule out other oculo-facial-digital and visceral defects which may be commonly associated with these disorders.
References
- Poretti A, Huisman TAGM, Scheer I, Boltshauser E (2011) Joubert syndrome and related disorders: Spectrum of neuroimaging findings in 75 patients. Am J Neuroradiol 32(8): 1459-1463.
- Van Beek EJR, Majoie CBLM (2000) Case 25: Joubert syndrome. Radiology 216(2): 379-382.
- Akhtar A, Hassan SA, Falah NU, Khan M, Sheikh FN, et al. (2019) Joubert Syndrome: A Rare Radiological Case. Cureus 11(12): 1-9.
- Parisi MA (2009) Clinical and molecular features of Joubert syndrome and related disorders. Am J Med Genet Part C Semin Med Genet 151(4): 326-340.
- Maria BL, Hoang KBN, Tusa RJ, Mancuso AA, Hamed LM, et al. (1997) “Joubert syndrome” revisited: Key ocular motor signs with magnetic resonance imaging correlation. J Child Neurol 12(7): 423-430.
- Poretti A, Boltshauser E, Loenneker T, Valente EM, Brancati F, et al. (2007) Diffusion tensor imaging in Joubert syndrome. Am J Neuroradiol 28(10): 1929- 1933.
- Gleeson JG, Keeler LC, Parisi MA, Sarah E Marsh, Phillip F Chance, et al. (2004) Molar tooth sign of the midbrain – hindbrain junction: occurrence in multiple distinct syndromes. Am J Med Genet A 125A(2): 125-134.
