 Research Article
Research ArticleAbstract
Background: The precursors for heme, 5-aminolevulinic acid (ALA) and porphobilinogen (PBG), are elevated in the urine of patients during an attack of one of the acute porphyrias. ALA and PBG can be measured directly by liquid chromatography tandem mass spectrometry (LC-MS/MS) without derivatization. However, ion suppression can severely reduce the sensitivity. The aim was to develop a robust LC-MS/MS method for measuring ALA and PBG without the need for derivatization.
Methods: In the method described here, silica Solid Phase Extraction (SPE) cartridges are used to remove the ion interferences from urine samples prior to LC-MS/MS. The isotopic internal standards, 13C2 15N-ALA and 13C2-PBG, were added to urine samples. The samples were then diluted in water, eluted through the silica SPE cartridges, diluted further with acetonitrile, and analysed by LC-MS/MS with a silica HPLC column.
Results: The ALA and PBG signals were significantly increased after elution of urine through silica SPE cartridges. The Coefficients of Variation (CVs) were typically < 8.5%, and recoveries ranged from to 107% to 112%. The lower limit of quantitation was 1.4 μmol/L for ALA and 1.0 μmol/L for PBG.
Conclusion: Ion interferences were difficult to separate from ALA and PBG using standard chromatography conditions. However, silica SPE cartridges were effective at removing ion suppression, therefore improving the sensitivity and reliability of the method.
Keywords: Acute Porphyria; 5-Aminolevulinic Acid; Ion Suppression; LC-MS/MS; Porphobilinogen; Silica SPE
Abbreviations: AIP:Acute Intermittent Porphyria; ALA: 5-Aminolevulinic Acid; CV:Coefficient of Variation; HILIC:Hydrophilic Interaction Chromatography; HPLC:High Performance Liquid Chromatography; LC-MS/MS:Liquid Chromatography- Tandem Mass Spectrometry; MRM:Multiple Reaction Monitoring; PBG: Porphobilinogen; SPE:Solid Phase Extraction; VP:Porphyria Variegata
Introduction
The precursors, 5-Aminolevulinic Acid (ALA) and Porphobilinogen (PBG), are involved in the heme biosynthetic pathway. ALA and PBG concentrations are elevated in urine and plasma during an attack of the acute porphyrias, such as Acute Intermittent Porphyria (AIP), or porphyria Variegata (VP) and are used as markers to screen for these conditions [1-4]. Urinary ALA can also be elevated in cases of lead poisoning [5,6].
ALA and PBG have previously been measured by colorimetric methods using ion chromatography and complexation with Ehrlich’s reagent [7,8], by High Performance Liquid Chromatography (HPLC) [9], and more recently by liquid chromatography mass spectrometry (LC-MS/MS) [1,2,10]. We previously measured ALA and PBG colorimetrically using a kitset method. However, colorimetric methods have lower sensitivity and specificity than mass spectrometry methods which can lead to falsely elevated results, especially at lower concentrations [1,11].
The main problem with measuring ALA and PBG in urine is loss of signal from ion suppression. Ion suppression is typically caused by the presence of non-volatile substances in the sample matrix that affect the ionization of the analytes and can dramatically decrease the sensitivity in mass spectrometry [12,13]. Urinary ALA and PBG have previously been measured by LC-MS/MS either directly [1] or with pre-column derivatization [10,14]. Benton et al. [1] described a method that reported to separate ALA and PBG in urine from ion interferences without the need for pre-column sample clean-up. However, their method required unorthodox chromatography conditions, including an injection volume of 100 μL and a high flow rate of 1 mL/minute through a 50 mm long HILIC HPLC column with a 2.1 mm internal diameter, as well as solvent splitting post column before entering the mass spectrometer [1]. These conditions could potentially overload the column and lower the life of the column. There is a need for a more robust LC-MS/ MS method to measure ALA and PBG directly using standard chromatography conditions that can be replicated in any clinical laboratory. We describe an efficient and reliable LC-MS/MS method for the direct measurement of urinary ALA and PBG that uses silica Solid Phase Extraction (SPE) cartridges to effectively address the problem of ion suppression.
Materials and Methods
Reagents
Acetonitrile was purchased from Merck (Darmstadt, Germany). Ammonium formate, 5-aminolevulinic acid, and porphobilinogen were purchased from Sigma (St Louis, MO, USA). 13C2 15N-5- Aminolevulinic acid (HCl) was obtained from BDG Synthesis (Wellington, NZ), and 13C2-porphobilinogen was purchased from IsoSciences (Ambler, PA, USA).
Standards
Calibration standards containing ALA and PBG in lyophilized urine were obtained from Recipe (Munich, Germany) and diluted in distilled water to make 6 calibration points for routine assays. Urine was also spiked with pure ALA and PBG (Sigma) to investigate the linear range of the assay. A working internal standard containing 100 μmol/L of both 13C2-PBG and 13C2 15N-5-ALA was prepared in distilled water.
Sample Extraction
In a 1.5 mL microcentrifuge tube, 500 μL of distilled water, 50 μL of urine, calibration standard, or control, and 10 μL of internal standard (containing 100 μmol/L 13C2 15N-ALA and 13C2-PBG) was added, and the samples were vortex mixed for 20 seconds. The samples were then eluted through Strata SI-1 silica (55 μm, 70 Å, 100 mg mL-1) SPE cartridges (Phenomenex, CA, USA). Fifty microlitres of the eluent was then extracted into 300 μL of acetonitrile. The samples were then vortexed, centrifuged at 13,000 × g for 5 minutes, then transferred to HPLC vials and capped for analysis.
LC-MS/MS
Samples were analysed using an Agilent 6490 mass spectrometer connected to an Agilent 1290 Infinity HPLC system. A Luna silica 50 x 3 mm, 3 μm (Phenomenex) with a silica guard column was used for the separation. The injection volume was 10 μL, and the injector wash solvent was from a wash vial containing distilled water. The column was maintained at room temperature (approximately 250oC). Mobile phase A consisted of 95% water and 5% acetonitrile containing 10 mmol/L ammonium formate. Mobile phase B contained 95% acetonitrile, 5% water, and 10 mmol/L ammonium formate. The flow rate was 0.3 mL/min and the flow was diverted to waste for the first 5 min of the gradient run. The gradient started at 7% A (93% B) and went to 10% A (90% B) over 3 min, then stepped to 20% A (80% B) at 3.1 min and stayed there until 6 min. Then the gradient stepped up to 50% A (50% B) at 6.1 min and went to 70% A (30% B) at 10 min. At 10.1 min the gradient went back to starting conditions 7% A (93% B) until 12 min. Mass spectrometry was carried out in positive ion mode using Electrospray Ionisation (ESI). The gas Temperature was 250oC, the gas flow rate was 14 l/min, the nebulizer pressure was 20 psi, the sheath gas flow rate was 11 l/min, the capillary voltage was 3000 V, and the nozzle voltage was set to 1500 V. The fragmentor voltage was set to 380 V, the cell accelerator voltage was set to 5 V, and the dwell times were set to 200 ms. ALA and PBG were detected using Multiple Reaction Monitoring (MRM). The resolution was set to enhanced for PBG and 13C2-PBG and unit for ALA and 13C2 15N-5- ALA. The MRM mass transitions for quantifier and qualifier ions and collision energies are shown in Table 1. To investigate the effect of ion suppression on the ALA and PBG signals, the same urine sample was compared with and without elution through silica SPE cartridges while undergoing the same dilutions. In order to evaluate the loss of ALA and PBG on the silica SPE cartridges, a 100 μmol/l aqueous standard containing no matrix interferences was both eluted through a silica SPE cartridge and not eluted through an SPE cartridge. The recovery of ALA and PBG was calculated by the ratio of the peak areas.
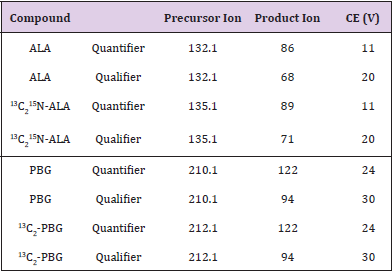
Table 1: Mass spectrometry parameters.
Method Validation
To evaluate precision and accuracy, a normal random urine sample was spiked with 0, 5, 50, and 200 μmol/l ALA (Sigma) and 0, 5, 20, and 50 μmol/L PBG (Sigma). Five replicates of each of the 4 different concentration levels were analysed for ALA and PBG over three separately calibrated runs. The low and a high lyophilised urine Quality Control (QC) material purchased from Recipe were analysed with each batch and the results were evaluated after 5 runs. The LC-MS/MS method was compared with ALA and PBG results obtained using the BioRad (CA, USA) colorimetric method for 10 urine samples with a varied concentration range from the Royal College of Pathologists of Australasia quality assurance program.
Results
LC-MS/MS
The use of silica SPE cartridges to remove the ion suppression significantly increased the size of the ALA and PBG peaks in urine samples, despite the silica solid phase removing some of the ALA and PBG (Figure 1). The recovery from a 100 μmol/L aqueous standard was 75% for ALA and 85% for PBG after elution through the SPE cartridges (Figure 2). With the dwell times set to 200 ms and the resolution set to “enhanced” for PBG and the internal standard (13C2-PBG), there was no detectable interference caused by the small mass difference (2 Da) or cross-talk in the PBG MRM after injecting 100 μmol/L 13C2-PBG. However, there was some minor interference (<1.5%) observed in the 13C2-PBG MRM when a high PBG standard (100 μmol/L) was injected.
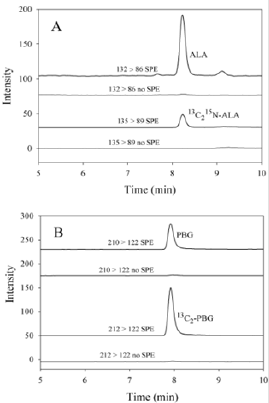
Figure 1:
(A) MRMs of ALA and 13C2 15N-ALA, and
(B) PBG and 13C2-PBG in a urine sample containing 89.9 μmol/l ALA, 14.7 μmol/l PBG and internal standard. PBG elutes at 7.9 min, and ALA elutes at 8.1 min The LC-MS/ MS conditions were the same for each injection.
Figure 2: The effect of SPE for 100 μmol/l aqueous ALA and PBG standards. The dashed line represents chromatograms from standards which have been through the silica SPE cartridges. The solid lines were standards that were not eluted through SPE cartridges but underwent the same dilutions. The mass transitions shown are 132 → 86 for ALA, and 210 → 122 for PBG.
Method Validation
Carry-Over and LLOQ: Carry-over was a problem for the measurement of PBG with an autosampler wash solvent containing high organic solvent. Therefore, the autosampler needle was rinsed from a wash vial containing distilled water. The Lower Limit of Quantitation (LLOQ) for ALA (defined as the concentration where the imprecision was < 20%) was 1.4 μmol/l. The LLOQ for PBG was 1.0 μmol/l.
Linearity: The r2 for the ALA calibration curve was 0.998 and the r2 for the PBG calibration curve was 0.999. Results from spiked urine showed that the assay was linear up to at least 360 μmol/l ALA, and 200 μmol/l PBG.
Precision and Accuracy: The between batch Coefficient of Variation (CV) for ALA ranged from 2.5% to 6.2%, the within batch CVs for ALA ranged from 2.5 to 8.4%, and the recoveries ranged from 110 to 112%. The between batch CVs for PBG ranged from 1.5% to 20% (near the LLOQ), the within batch CV for PBG ranged from 2.3% to 4.1%, and the recoveries ranged from 107 to 110% (see Table 2). CVs below 15%, and recoveries of 100% (±15%) were considered acceptable. The Recipe QCs were all within the acceptable ranges provided by the manufacturer. Over 5 separate runs, the ALA low QC mean was 30.4 μmol/L (ref value 32.3 (acceptable range 24.2 – 40.4) μmol/L), the high ALA QC was 131.5 μmol/L (ref value 142 (113 – 170) μmol/L), the low PBG QC was 6.5 μmol/L (ref value 7.9 (5.5 – 10.3) μmol/L), and the high PBG QC was 74.8 μmol/L (ref value 69.7 (52.3 - 87.1) μmol/L). The between batch CVs for the Recipe controls were < 3.2% for ALA and < 5.2% for PBG.
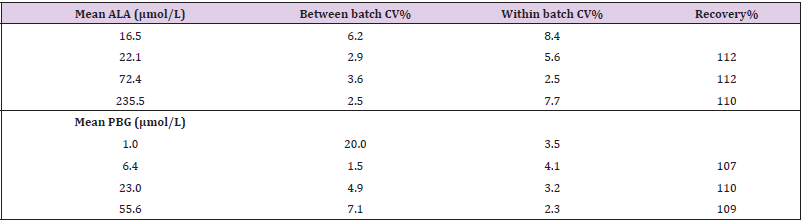
Table 2: Precision and accuracy for ALA and PBG.
Method Comparison: The results comparing the LC-MS/MS method with the colorimetric method are shown in Table 3. There was a strong correlation between the methods for both ALA (r2 = 0.994) and PBG (r2 = 0.985). Passing-Bablok analysis indicated that the ALA (y = 1.098x – 0.1) results had a positive bias for LC-MS/ MS but were not significantly different for PBG (y = 0.962x - 0.2) compared to the colorimetric method.
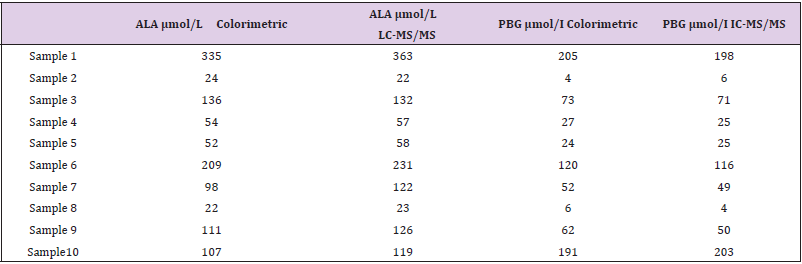
Table 3: Method comparison.
Discussion
There are few reported methods for the measurement of ALA and PBG in urine by LC-MS/MS. Most of these methods involve sample clean-up and/or derivatization to deal with the problem of ion suppression. Derivatization methods require extensive sample preparation [10,14]. Zhang et al. performed SPE using Waters Oasis HLB polymer reversed phase cartridges, and butylated ALA and PBG prior to analysis by LC-MS/MS [10]. Floderus et al. [14] derivatized the amino groups on ALA and PBG with phenylisothiocyanate to measure them in serum by LC-MS/MS. Ford et al. [15] measured PBG by LC-MS/MS without the need for derivatization, but performed a sample clean-up step with Waters Oasis HLB polymeric reversed phase SPE cartridges. While Benton et al. [1] separated ALA and PBG from the matrix interferences chromatographically, we found that when using standard chromatography conditions, ion suppression still hindered the detection of ALA and PBG, and some form of pre-column sample clean-up was required. Relying on the HPLC column to separate analytes from the urine matrix interference can make the method less reliable because small changes to the chromatography may lead to a severe loss in signal for ALA and PBG. The commercially available isotopes, 13C2 15N-5- aminolevulinic acid (HCl) (13C2 15N-ALA) and 13C2-porphobilinogen (13C2-PBG), were used as internal standards in the current method. Ford et al. also used 13C2-PBG as the internal standard to measure PGB [15], and Zhang et al. [10] used the isotopic internal standards 13C2-PGB and 13C5 15N-ALA for the analysis of ALA and PBG by LCMS/ MS. The internal standard used by Benton et al. [1] (6-amino- 5-oxohexanoic acid) is not readily available and is structurally different to both ALA and PBG.
Comparing the mass spectrometer signal for a biological sample such as urine with an aqueous standard is an effective way to investigate the effect of ion suppression from the sample matrix on the sensitivity [13]. The peak areas for both ALA and PBG were much lower without removing the interfering substances in the sample matrix. However, this interference was effectively removed by elution of diluted urine in an aqueous phase through silica SPE cartridges prior to LC-MS/MS. This suggests that the ion interference present in urine is likely to be caused by strongly hydrophilic cations such as sodium and potassium. ALA and PBG also have an affinity for silica, and this has been demonstrated by the use of a silica HPLC column for the chromatographic separation. However, in a completely aqueous medium, ALA and PBG are not well retained by the silica SPE cartridges and mostly elute straight through, whereas the ion interferences in urine appear to have a stronger affinity for silica and are effectively removed from the samples. The apparent analyte loss (of approximately 25% of ALA, and 15% of PBG) during sample clean-up is relatively minor compared to the loss in signal from ion suppression. To account for these analyte losses, the assay relies on the use of isotopic internal standards for reliable quantitation.
Conclusion
The LC-MS/MS method described here measures ALA and PBG in urine directly without the need for pre-column derivatization, uses commercially available isotopic internal standards, and does not suffer from low sensitivity due to matrix interferences. This method is relatively simple and produces results with acceptable accuracy and precision, and can be employed in any laboratory with LC-MS/MS to measure urinary ALA and PBG for aiding the diagnosis of porphyrias such as AIP and VP.
Declarations of Interest
None.
References
- Benton CM, Couchman L, Marsden JT, Rees DC, Moniz C, et al. (2012) Direct and simultaneous determination of 5-aminolaevulinic acid and porphobilinogen in urine by hydrophilic interaction liquid chromatography-electrospray ionisation/tandem mass spectrometry. Biomed Chromatogr 26(8): 1033-1040.
- Benton CM, Couchman L, Marsden JT, Rees DC, Moniz C, et al. (2013) Direct and simultaneous determination of 5-aminolaevulinic acid and porphobilinogen in human serum or plasma by hydrophilic interaction liquid chromatography-atmospheric pressure chemical ionization/tandem mass spectrometry, Biomed. Chromatogr 27(2): 267-272.
- Stein PE, Badminton MN, Rees DC (2017) Update review of the acute porphyrias. Br J Haematol 176(4): 527-538.
- Sardh E, Harper P, Andersson DE H, Floderus Y (2009) Porphobilinogen as a sensitive biomarker to monitor the clinical and therapeutic course of acute intermittent porphyria attacks. Eur J Int Med 20(2): 201-207.
- Davies JR, Andelman SI (1967) Urinary delta-aminolevulinic acid (ALA) levels in lead poisoning, I. A modified method for rapid determination of urineary delta-aminolevulinic acid using disposable ion-exchange chromatography columns. Arch. Environ. Health 15(1): 53-59.
- Davis JR, Abrahams RH, Fishbein WI, Fabrega EA (1968) Urinary delta-aminolevulinic acid (ALA) levels in lead poisoning, II. Correlation of ALA values with clinical findings in 250 children with suspected lead ingestion, Arch. Environ. Health 17(2): 164-171.
- Watson CJ, Schwartz S (1941) A simple test for urinary porphobilinogen. Proc Soc Exp Biol Med 47(2): 393-394.
- Mehani S (1964) A rapid method for the determination of delta amino-laevulinic acid in urine. Brit J Industr Med 21(1): 78-80.
- Ho J, Guthrie R, Tieckelmann H (1986) Detection of δ-aminolevulinic acid, porphobilinogen and porphyrins related to heme biosynthesis by high-performance liquid chromatography. J Chromatogr 375(1): 57-63.
- Zhang J, Yasuda M, Desnick RJ, Balwani M, Bishop D, et al. (2011) A LC–MS/MS method for the specific, sensitive, and simultaneous quantification of 5-aminolevulinic acid and porphobilinogen. J Chromatogr 879(24): 2389-2396.
- Verstraeten L, Ledoux MC, Moos B, Callebaut B, Cornu G, et al. (1922) Interference of Tienam in colorimetric determination of 5-aminolevulinic acid and porphobilinogen in serum and urine. Clin Chem 38(12): 2557-2558.
- Annesley TM (2003) Ion suppression in mass spectrometry. Clin Chem 49(7): 1041-1044.
- Pitt JJ (2009) Principles and applications of liquid chromatography-mass spectrometry in clinical biochemistry. Clin Biochem Rev 30(1): 19-34.
- Floderus Y, Sardh E, MÖller C, Andersson C, Rejkjaer L, et al. (2006) Variations in porphobilinogen and 5-aminolevulinic acid concentrations in plasma and urine from assymtomatic carriers of the acute intermittent porphyria gene with increased porphyrin precursor excretion. Clin Chem 52(4): 701-707.
- Ford RE, Magera MJ, Kloke KM, Chezick PA, Fauq A, et al. (2001) Quantitative measurement of porphobilinogen in urine by stable-isotope dilution liquid chromatography-tandem mass spectrometry. Clin Chem 47(9): 1627-1632.
