 Case Report
Case ReportAbstract
Introduction: We present a case report of self-curing congenital reticulumhistiocytosis (Hashimoto-Pritzker disease), a rare disease, with an annual incidence estimated in 2 to 9 cases per million children.
Case Report: At birth, she had multiple papular-nodular, erythematous lesions, some covered by crusts, on her body. The lesions completely disappeared at 7 weeks of age.
Discussion: The real incidence of Hashimoto-Pritzker’s disease is slightly higher than that observed in the literature, being underestimated due to the complex clinical diagnosis and the rapid and spontaneous resolution of the disease.
Introduction
Langerhans cell histiocytoses encompass several diseases, with a wide spectrum of forms of presentation and evolution [1]. Among all variables, there are similarities between then, such as the presence of clonal proliferation of Langerhans cells with Birbeck granules and positive S100 and CD1a antibodies in the immunohistochemistry test [1-3]. Self-curing congenital reticulumhistiocytosis, also known as Hashimoto-Pritzker disease, of benign course, is characterized by the presence of cutaneous lesions at birth or in the neonatal period, with spontaneous resolution and absence of systemic manifestations in most cases. In rare exceptions, it is possible to spread or worsen the disease [1].
Case Report
Preterm newborn, born at 36 weeks and 3 days of gestation, female, with normal delivery. At birth, weighing 2,270 grams, 44.5 cm in height, 35 cm head circunference, 29 cm chest circunference, in addition to an Apgar Scale of 9 in the first minute and 10 in the fifth minute. The newborn’s mother was colonized by group B Streptococcus (positive vaginal / rectal swab), being allergic to penicillin, having used clarithromycin in the prepartum period. In the dermatological examination at birth, we presented several nodular, erythematous lesions, some recovered by crusts on the face, upper limbs, trunk, abdomen and lower limbs, in addition to erythema and desquamation on the soles of the feet (Figures 1 & 2).
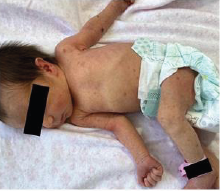
Figure 1: Erythematous papules with crusts on the trunk and limbs.
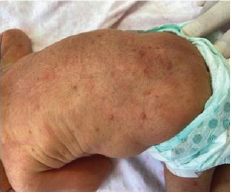
Figure 2: Erythematous papules covered by nodular crusts.
She had a normal routine neonatal laboratory evaluation and negative serology for the TORCHS group. The anatomopathological examination of cervical lesions shows the proliferation of epithelioid cells in the dermis, with broad cytoplasm and rhiniform nuclei, or multinucleate, accompanied by a moderate inflammatory infiltrate predominantly interstitial and perivascular lymphocyte, without observing atypical mitosis or necrosis (Figure 3). The immunohistochemical study showed a positivity of the accessories S100, CD68 and CD1a, being compatible with clinical histological aggregates, such as Langerhans cells (Figure 4).
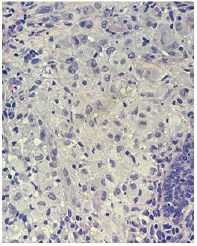
Figure 3: Histopathological examination of the skin showing dense infiltrate of histiocytes in the dermis with ample cytoplasm and rhiniform nuclei. (HE, 100x magnification).
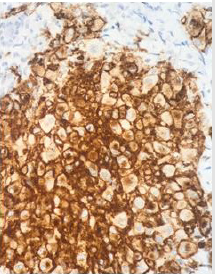
Figure 4: Positive immunohistochemistry for the CD1a antigen.
Returning to the clinic at 2 months and 2 days old, the patient’s mother reported complete disappearance of the lesions when the child was 7 weeks old. Upon dermatological examination, the patient no longer had skin lesions (Figures 5 & 6). It was therefore concluded that it was a self-curing congenital reticulumhistiocytosis, or also known as Hashimoto-Pritzker disease.The patient is undergoing regular outpatient dermatological follow-up.
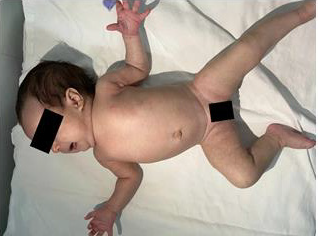
Figure 5:
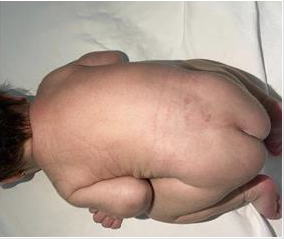
Figure 6:
Discussion
Langerhans cell histiocytosis is a rare disease, with an estimated annual incidence of 1 to 2 cases per million adults and 2 to 9 cases per million children [4]. It is a heterogeneous condition: it may present as a single episode or with recurrences, affect one or more organ systems and manifest itself in any age group, with the neonatal period being the most common. Although it is proven that the disease occurs by the clonal expansion of the myeloid dendritic Langerhans cells, its etiology still remains uncertain, requiring anatomopathological examination to confirm the diagnosis. Among Langerhans cell histiocytoses, self-curing congenital reticulohistiocytosis or Hashimoto-Pritzker disease stands out, which was initially described by Hashimoto and Pritzker in 1973. This is characterized by the presence of benign skin lesions such as papules, nodules or vesicles, in most cases not having systemic involvement. The course of the disease is usually self-limited: it appears at birth or in the neonatal period, and usually regresses in the first year of life, without treatment [5]. Despite the reports regarding the benign and self-limited nature of the disease, there are no absolute criteria that can reliably distinguish autoregressive from non-auto-regressive forms in the neonatal period and early childhood [6,7]. In addition, histopathological and immunohistochemical criteria have been addressed in several studies in the literature and have not shown a significant difference between the findings of patients with the limited form and the systemic form.
Many experts believe that the real incidence of Hashimoto- Pritzker’s disease is slightly higher than that observed in the literature, being underestimated due to the complex clinical diagnosis and the quick and spontaneous resolution of the disease. For the prompt diagnosis, we must consider the existence of clinical lesions together with histopathological findings: presence of histiocytes with Birbeck granules in electron microscopy, and CD1a antibodies in immunohistochemistry [8]. In this context, it is essential to pay attention to the possibility of other exclusion diagnoses, such as neonatal lupus, drug rash, herpes simplex vírus infection, neonatal toxic erythema, melanosis pustular and eosinophilic pustular folliculitis.
Although the disease appears harmless, it is necessary to perform careful screening to analyze other possible systemic disorders, in addition to the skin - a situation that occurs in approximately 40% of diagnosed newborns4. In this case, it mainly affects the mucous membranes, bones and pituitary gland. In addition, it is important to emphasize that even in the absence of systemic involvement in the initial course of the disease, there is a possibility of progression to this or even of clinical relapse with the recurrence of skin lesions. There is no specific treatment for Hashimoto-Pritzker disease. The recommended approach is to wait for spontaneous regression of the lesions and perform periodic outpatient follow-up. In case of persistent skin lesions, the use of topical corticosteroids or topical nitrogen mustard can be effective 1,3. Due to reports of recurrence, patient follow-up must be strict and long-term for early detection of possible systemic spread of the disease.
Acknowledgement
None.
Conflict of Interest
No conflict of interest.
References
- Pan Y, Zeng X, Ge J, Liu X, Chen Y, et al. (2019) Congenital self-healing langerhans cell histiocytosis: clinical and pathological characteristics. Int J Clin Exp Pathol 12(6): 2275-2278.
- Lee YH, Talekar MK, Chung CG, Bell MD, Zaenglein AL (2014) Congenital self-healing reticulohistiocytosis. J Clin Aesthet Dermatol 7(2): 49-53.
- Orle J, Mósca AM, Sousa MA, Lima CMO, Adriano AR, et al. (2011) Congenital self healingreticulohistiocytosis in a newborn (Hashimoto Pritzker). A Bras Dermatol 86(4): 785-788.
- Briner RMS, Fierling R, Gerber NU, Kamarachev J, Arlettaz R (2018) Hashimoto- Pritzker Langerhans cell histiocytosis in a neonate. Cheseau-sur-Lausanne: Swiss Society of Neonatology :1-19.
- Mandel VD, Ferrari C, Cesinaro AM, Pellacani G, Del Forno C (2014) Congenital “self-healing” Langerhans cell histiocytosis (Hashimoto-Pritzker disease): A report of two cases with the same cutaneous manifestations but different clinical course. Journal of Dermatology 41: 1098-1101.
- Battistella M, Fraitag S, Teillac DH, Brousse N, De Prost Y, et al. (2010) Neonatal and early infantile cutaneous Langerhans cell histiocytosis: comparison of self-regressive and non-self-regressive forms. Arch Dermatol 146: 149-156.
- Kapur P, Erickson C, Rakheja D, Carder KR, Hoang MP (2007) Congenital self-healing reticulohistiocytosis (Hashimoto-Pritzker disease): ten-year experience at Dallas Children’s Medical Center. J Am Acad Dermatol 56:290-294.
- Quattrino AL, Silveira JCG, Diniz C, Briggs MC, Vilar E (2007) Histiocitose de células de Langerhans: relato de caso e revisão da literatura. Anais Brasileiros de Dermatologia 82(4): 337-341.
