 Research Article
Research ArticleAbstract
Unfolded Protein Response (UPR) signaling as a result of ER stress has been recently implicated in apoptotic and autophagic pathways. Given its potentially critical role, we investigated this interaction in a pathogenic infection, a common source of ER stress. Trichomonas vaginalis is an extracellular parasite responsible for the most common sexually transmitted disease in the world. By analyzing protein expression dynamics in cervical epithelial cells infected with T. vaginalis, we identified a strong induction of UPR markers GP378/BiP and p-elF5a, as well as pro-apoptotic proteins such as Bim and cleaved caspases, indicating a marked activation of apoptosis. Conversely, pro-survival signaling molecules Mcl-1 and Bcl-XL and autophagy molecules LC3-II and p62 showed transient inductions. How UPR dynamics may contribute to the sustained expression of pro-apoptotic BH3-only proteins to shift the balance of survival/cell death to the side of apoptotic cell death requires further investigation.
Keywords: Unfolded protein response; endoplasmic reticulum (ER) stress; p-elF5a; Trichomonas vaginalis; Autophagy; Apoptosis
Abbreviations: UPR: Unfolded Protein Response; ER: Endoplasmic Reticulum; SDS-PAGE: Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis; PVDF: Polyvinylidene Difluoride; SD: Standard Deviation; ASK: Apoptotic-Signaling Kinase
Introduction
Trichomonas vaginalis is an extracellular protozoan parasite known to cause vaginitis and urethritis. Trichomoniasis is the most common non-viral sexually transmitted disease in the world with more than 170 million cases reported per year [1,2]. T. vaginalis colonizes at the epithelium of the human genital tract and is transported into cells by hydrolyzing organic components into glucose, leading to premature membrane rupture, preterm delivery, and low birth weight. Trichomoniasis has also been associated with atypical pelvic inflammatory disease, infertility, and increased susceptibility to HIV infection and invasive cervical cancer [3-7].
Trichomonas vaginalis is an understudied parasitic organism whose pathogenic mechanisms remain unclear, even though it is the cause of a major public health problem. Current literature has focused its contact-dependent mechanisms, where the parasite adheres to vaginal epithelial cells to induce cytotoxicity, as a major contributing factor towards disease development [7-13]. As for contact-independent mechanisms, it is well known that the secretion of virulence factors by parasitic protists plays a fundamental role in multifactorial host-parasite interactions. As such, other studies have highlighted the effector proteins and proteases that are secreted by Trichomonas vaginalis into the urogenital tract as central players in pathogenesis [14]. We, however, focus on the subsequent cellular stress events following these mechanisms in SiHa cells, a carcinoma cell line of the cervix.
Cellular stress plays a key role in both of these mechanisms. It can be induced by external or internal cues and activates several well-orchestrated processes aimed at either restoring cellular homeostasis or committing to cell death [15-17]. One of these processes is the Endoplasmic Reticulum (ER) stress response. The Endoplasmic Reticulum (ER) is a highly dynamic network composed of sac-like structures and tubules. The major function of the ER is to fold and mature nascent proteins that transit through the secretory pathway [18]. Perturbations at the cellular level can affect ER homeostasis and accumulate unfolded proteins in the ER lumen, a condition referred to as ER stress. To alleviate ER stress and ensure cell survival, the Unfolded Protein Response (UPR) promotes degradation of misfolded proteins and induces greater protein folding activity in the ER lumen. However, under prolonged conditions, cells that become irreversibly damaged are subjected to autophagy and ultimately eliminated by apoptosis [19-22]. In ER stress-mediated cell death, transcriptional and posttranscriptional mechanisms are activated to regulate pro-apoptotic members of the Bcl-2 family to facilitate cytochrome-c release from the mitochondria and calcium release from the ER to engage downstream apoptotic signaling events [23, 24].
Notably, mitochondria-dependent apoptosis has been heavily implicated in T. vaginalis-induced pathogenesis. One study identified the release of cytochrome c into the cytosol, the activation of caspase-3 and caspase-9, and the cleavage of PARP following T. vaginalis infection [25-27]. T. vaginalis infection of human neutrophils also resulted in apoptosis through the activation of caspase-3 and the reduction of Mcl-1 expression [27]. Additional studies have argued the importance of the balance of Bcl-2 family proteins in T. vaginalis-mediated cell death [25]. Bcl- 2 proteins are grouped into three classes based on their activities and the particular Bcl-2-homology domains they contain: antiapoptotic Bcl-2 proteins (Bcl-2, Mcl-1, and Bcl-xL), pro-apoptotic multidomain proteins (Bak and Bax), and BH3-only pro-apoptotic proteins (Bad, Bid, Puma, and Bim) [28]. In RAW264.7 cells, T. vaginalis induced apoptosis through the action of Bcl-xL but not that of Bcl-2 [29]. These findings merit further investigation on the balance between pro-apoptotic and anti-apoptotic Bcl-2 proteins during T. vaginalis infection.
Furthermore, many intracellular pathogens, such as protozoan parasites belonging to genus Plasmodium, Leishmania, Toxoplasma and Cryptosporidium, are known to induce an UPR activity in infected cells [30]. However, UPR pathways in cells infected by extracellular parasites have yet to be reported. T. vaginalis-induced apoptosis may be mediated by UPR. The UPR is mediated by three ER-resident transmembrane proteins that can act as ER stress sensors, IRE1, PERK and ATF6, which are in turn regulated by BiP (also known as glucose regulated protein 78 or GRP78), a major chaperone in the ER lumen [17-20,22-24]. We hypothesized that T. vaginalis-infected cervical epithelial cells would demonstrate altered expression patterns of these key molecules as well as significantly affect downstream pro-apoptotic products such as JNK phosphorylation or CHOP expression and phagophore-promoting events including LC3 lipidation which then binds to the phagophore to form autophagosomes. Understanding how T. vaginalis infection regulates mechanisms such as ER stress, autophagy, and apoptosis is critical for the protection of host cells. In this study, we identify the induction of ER stress in SiHa cells by T. vaginalis infection, which ultimately sequentially resulted in the autophagy and apoptosis of the SiHa cells.
Materials and Methods
Antibodies and reagents
The following antibodies were used : anti-LC3(L8918), anti-p62(P0067) from Sigma-Aldrich (St. Louis, Missouri, USA), anti-BiP (GRP78) (#3177), anti-phospho-eIF2α (#3597), antiphospho- ERK1/2 (#4370), anti-ERK1/2 (#4695), anti-phospho- SAPK/JNK (#9251), anti-SAPK/JNK (#9258), anti-phospho-p38 MAPK (#9216), anti-p38 MAPK (#9212), anti-Atg4B (#5299), anti-Atg5 (#8540), anti-Atg7 (#2631), anti-PARP (#9532), anti- Bcl-xL (#2764), anti-Mcl-1 (#5453), anti-Bim (#2933), anti- Cleaved caspase-3 (#9601), anti-Cleaved caspase-9 (#9501), anti- Bak (#6947), anti-Bax (#5023) from Cell signaling Technology (Burlingame, CA, USA), anti-α-tubulin (sc-32293), anti-actin (sc- 1616-R) from Santa Cruz Biotechnology (Dallas, TX, USA).
Host cell culture
Human cervical epithelial cancer cell lines (SiHa cells) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). SiHa cells were maintained under 5% CO2 at 37℃ in Dulbecco’s Modified Eagle’s Medium (DMEM) with 10% heat-inactivated fetal bovine serum (FBS, Gibco BRL, Grand Island, NY, USA) and 1X Antibiotic-Antimycotic (Anti-Anti, Gibco BRL).
Trichomonas vaginalis culture
The T. vaginalis T106 isolate obtained from one of the author Prof. Jae-Sook Ryu was provided by Prof. J. K. Alderete. T. vaginalis T106 isolate was cultured in TYM medieum, which contained 20% (w/v) Tryticase peptone, 10% (w/v) Yeast extract, 5% (w/v) Moltose monodydrate, 1% (w/v) L-cystein, 1% (w/v) Ascorbic acid, 1% (w/v) K2HPO4, 1% (w/v) KH2PO4 (pH 6.2) with 10% heatinactivated horse serum (Sigma-Aldrich, St Louis, MO, USA) and Penicillin-Streptomycin (Gibco BRL) under 5% CO2 at 37℃. After 0.22-μm-pore filteration, the medium was used for culture. Live T. vaginalis was infected to SiHa cells at Multiplicities of Infection (MOI) of 1 for time-dependent in mixed-medium (DMEM(without Antibiotic):TYM = 2:1).
TCA Precipitation
After cells were washed with PBS (pH 7.4), cells were suspended in ice-cold 10% (w/v) TCA, incubated on ice for 30 min, scraped into microfuge tube, and centrifuged at 13,000 rpm for 5 min at 4℃. The resulting pellet was washed with ice-cold 5% (w/v) TCA and centrifuged. The supernatants were removed, and the pellets were washed with ice-cold acetone and then centrifuged. The pellets were resuspended in a solution of 0.1 M Tris-HCl (pH 11.0), 3 % SDS and sonicated. After centrifugation at 12,000 rpm for 2 min the supernatant was transferred to a new tube. 5X SDS-PAGE sample buffer was added to the sample and boiled for 5 min. Samples were be loaded SDS-PAGE gel or stored at -20℃.
Western Blot analysis
Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE) and western blot analysis were carried out to analyze the expression of various proteins. TCA precipitated-cells were loaded to SDS-PAGE gels for equal amounts, and then transferred to Polyvinylidene Difluoride (PVDF) membrane (Bio-Rad Laboratories, Hercules, CA, USA). The membranes were blocked with 5% skim milk in Tris-buffered saline containing 1% Tween 20 (TBST) and were incubated with various primary antibodies overnight at 4℃. Membranes were washed three times with TBST and then incubated with horseradish peroxidase-conjugated antirabbit and anti-mouse IgG (Santa Cruz Biotechnology) for 2 h at room temperature. After being washed three times, the blots were developed using ProNATM ECL Ottimo Western Blot Detection Kit (Trans Lab, Daejeon, Korea).
Statistical Analysis
Results were repeated at least three independent times and expressed as the mean ± Standard Deviation (SD). P values between groups were determined by a two-tails paired Student t tests. P < 0.05 was considered statistically significant.
Results
Induction of Endoplasmic Reticulum (ER) stress in SiHa cells by T. vaginalis infection
Given that ER stress has been reported to result from pathogenic stress signals such as infections, we examined the involvement of the UPR signaling pathway in T. vaginalis infection of SiHa cells. GRP78 (BiP) and phospho-eIF2α expression levels have been recognized as good monitors of UPR activation. Thus, to assess the involvement of ER stress, we measured the expression of phosphoeIF2α and GRP78 in T. vaginalis-infected SiHa cells.
The expression level of GRP78 was significantly increased 12 hrs after T. vaginalis MOI 1 infection and remained enhanced at 48 hrs (Figure 1A). Phospho-eIF2α expression was also significantly increased at 12 hrs after T. vaginalis infection and continued to increase up to 48 hrs (Figure 1A). These results demonstrate that representative markers of UPR activity were induced early-on in SiHa cells infected with T. vaginalis and continued to be highly expressed 48 hrs following time of infection.
Induction of autophagy in the SiHa cells infected with T. vaginalis
Since autophagy has been reported to be linked to infections and ER stress, we then investigated how the autophagy process is involved in the physiology of T. vaginalis-infected SiHa cells. As an indicator of autophagosome formation and autophagy, LC3 was detected in two forms: LC3-I and II (PE-conjugated form).
We found that the expression of the LC3-Ⅰ band was induced at 12 hrs (Figure 1B). On the other hand, LC3-Ⅱ expression significantly increased at 18 h, peaked at 24 hrs, and decreased at later time points (Figure 1B). ATG5 expression, which is important for phagophore formation, steadily increased in T. vaginalis infected SiHa cells while ATG4B and ATG7 expression levels were not significantly altered (Figure 1C). These results suggest that T. vaginalis infection may induce autophagy in SiHa cells.
The unique LC3-II expression dynamics prompted an examination of whether autophagic flux is affected by T. vaginalis infection. The dynamics of p62 (SQSTM1/sequestosome 1) protein expression was examined, because as a ubiquitin- and LC3-interacting protein, p62 binds and transports ubiquitinated proteins into autophagosomes for degradation. Expression levels of this autophagy substrate were significantly enhanced between 18 and 24 hrs and degraded after 36 hrs (Figure 1D). This finding suggests that clearance of ubiquitinated proteins by p62, and thus autophagic flux, was somehow blocked at 18–24 hrs following the infection and resolved later.
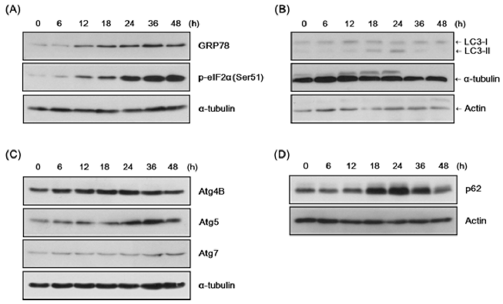
Figure 1: Induction of endoplasmic reticulum (ER) stress and autophagy in the SiHa cells infected with T. vaginalis infection.
(A) SiHa cells were infected with live T. vaginalis trophozoites at a multiplicities of infection (MOI) of 1 in mixed-medium (DMEM(without Antibiotic):TYM = 2:1) for the indicated times. Reactions were stopped by TCA precipitation. Cell lysates were prepared and subjected to immunoblot analysis using anti-GRP78, anti-phospho-eIF2α, and anti-α–tubulin antibodies.
(B) Samples were prepared as in (A) and analyzed with anti-LC3 antibody, anti-α-tubulin, and anti-actin antibody. Positions of LC3 I and LC3 II are indicated.
(C) Cell lysates were prepared as in (A) and analyzed with anti-Atg4B, anti-Atg5, anti-Atg7, and anti-α-tubulin anti-bodies.
(D) Samples were prepared as in (A) and analyzed with anti-p62 (SQSTM1/sequestosome 1) and anti-actin antibodies.
Induction of apoptosis the SiHa cells infected with T. vaginalis
The prolonged enhancement of GRP78 and phospho-eIF2α expression suggests that T. vaginalis infection induces continuous ER stress in SiHa cells. Furthermore, p62 protein expression dynamics indicate a change in autophagic flux following infection. These findings indicate the possibility of apoptosis pathway activation in T. vaginalis-infected SiHa cells. Anti-apoptotic and proapoptotic protein expression levels were analyzed in T. vaginalisinfected SiHa cells by western blot. Anti-apoptotic proteins such as Bcl-xL and Mcl-1 were significantly induced at 12 hrs and remained enhanced until 36 hrs (Figure 2A). However, the proapoptotic protein Bim, especially BimEL, was strongly induced from 18 to 48 hrs (Figure 2B). Another pro-apoptotic protein Bak demonstrated dynamics similar to Bim. The pro-apoptotic protein Bax also showed slight enhancement during the infection period (Figure 2B). Together, there is an early but brief induction of antiapoptotic proteins such as Bcl-xL and Mcl-1 by T. vaginalis infection, while pro-apoptotic proteins such as Bim and Bak were induced at later timepoints and strongly sustained. These data suggest the dominance of pro-apoptotic processes in infected SiHa cells.
Mitochondria-dependent apoptosis was analyzed by measuring protein expression levels of PARP and caspases 9 and 3. Cleaved caspase-9 and cleaved PARP were markedly elevated at later time points 36 and 48 hrs (Figure 2C), indicating that apoptosis was induced in T. vaginalis infected-SiHa cells. Interestingly, cleaved caspase-3 was induced early on at 18 hrs following infection (Figure 2C). When the activities of the MAPK signaling pathways were examined, p-ERK was dominant at early time points while p-JNK and p-p38 were induced and sustained strongly at later time points (figure 2D). These results suggest that JNK and p38 pathways are heavily involved in maintaining stress signals in SiHa cells infected with T. vaginalis.
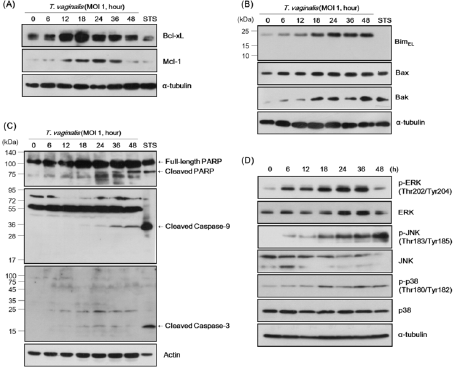
Figure 2: Induction of apoptosis and MAPK signaling events in SiHa cells by T. vaginalis-infection.
(A) SiHa cells infected with Live T. vaginalis trophozoites at MOI 1 in mixed-medium (DMEM(without Antibiotic):TYM = 2:1) were stopped by TCA precipitation. Staurospoine (STS, 1 μM, 16 h) was treated for positive control as a inducer of apoptosis. Cell lysates were prepared through the procedure described in the materials and methods section and then subjected to immunoblot analysis using anti-Bcl-xL, anti-Mcl-1, and anti-α-tubulin antibodies.
(B) Total cell lysates were prepared as in (A) and subjected to analysis using anti-Bim, anti-Bak, anti-Bax, and anti-α–tubulin antibodies.
(C) Samples were prepared like in (A) and subjected to immunoblot analysis using anti-PARP, anti-cleaved caspase-9, anticleaved caspase-3, and anti-actin antibodies. Positions of cleaved PARP, cleaved caspase-9, and cleaved caspase-3 are indicated.
(B) Cell lysates were prepared as in (A) and subjected to analysis using anti-phospho-ERK, anti-ERK, anti-phospho-JNK, anti- JNK, anti-phospho-p38, anti-p38, and anti-α–tubulin antibodies.
Discussion
Trichomonas vaginalis is an anaerobic, aerotolerant pathogen known to cause trichomoniasis, the most common sexuality transmitted disease in the world. Previous studies have implicated the importance of ER stress dynamics in pathogen-induced cell death. Here, we examine various protein expression patterns in cervical epithelial cancer (SiHa) cells to argue that T. vaginalis also utilizes ER stress pathways to promote disease. Specifically, we demonstrate that there is a differential induction of UPR signaling, autophagy, and apoptosis pathways. Affirming the induction of ER stress following T. vaginalis infection, we found that both GRP78 and phospho-eIF2α expression levels were significantly enhanced early on (12 hrs) and sustained at 48 hrs (Figure 1A). GRP78 is an important regulator of ER stress sensors. It can bind to IRE1, PERK, and ATF6 to inhibit their signaling under normal conditions. When ER stress is enhanced, GRP78 is released and these stress sensors can change conformation to their active states. Released GRP78 can then bind to misfolded proteins while activated IRE1, PERK, and ATF6 upregulate GRP78 production [16,19,21] making GRP78 a strong marker for ER stress.
Activated PERK can enhance autophagic activity by phosphorylating eIF2α to lead to LC3 lipidation, which then binds to the phagophore to form autophagosomes [16,17,19,21]. The increase in phospho-eIF2α expression between 24 and 48 hrs suggests an upregulation of this PERK/p-eIF2α/ATF4 pathway. In addition to lipidation of LC3, PERK induces the apoptosis through increase of CHOP expression. ATF4 and C/EBP homologous protein (CHOP), a transcription factor induced by ATF4, are known to transcriptionally regulate more than a dozen ATG genes [16,17,19,21]. In line with these pathways, we found an early increase in expression of LC3-Ⅱ and ATG5, suggesting the induction of autophagy in SiHa cells (Figure 1B, C). Interestingly, the expression of p62 was only briefly enhanced at 18-24 hrs and then resolved. p62 is responsible for transporting ubiquitinylated proteins into autophagosomes for degradation, suggesting that the autophagic flux was blocked for a short period following infection (Figure 1D).
PERK induced-apoptosis is regulated via inhibition of antiapoptotic Bcl-2 members and activation of pro-apoptotic BH3 proteins [17,21,24]. We found that pro-apoptotic factor Bim, as well as key cell death executioner proteins Bax and Bak, showed a strong and sustained induction at later time points. Compared to the sustained strong expression of p-elF2a, Bim, and Bak, autophagy indicator proteins LC3-Ⅱ and ATG5 showed only transient induction, suggesting that even though ER stress-induced autophagy serves as a pro-survival mechanism, cell death-promoting branches of the ER stress pathways prevailed. Relatively higher enhancement of pro-apoptotic proteins Bim and Bak compared to pro-survival BCLXL and Mcl-1 at late time points may also contribute to cell death [24,28,31]. Expression of the cleaved forms of Caspase-9 and PARP indicate a mitochondria-dependent apoptotic mechanism [16,17,21].
IRE1 is also known to contribute to both the autophagic and apoptotic pathways, specifically by promoting Jun-N-Terminal Kinase (JNK) phosphorylation [32]. Our data show that JNK and p38 were phosphorylated early following T. vaginalis infection and continued to be phosphorylated, demonstrating particularly strong expression after 18 hrs (Figure 2D). Phosphorylated JNK can in turn phosphorylate Bcl-2, allowing the dissociation of Bcl- 2 from Beclin-1 and the initiation of autophagy [15,17]. IRE1α also stimulates activation of the Apoptotic-Signaling Kinase-1 (ASK1), which activates downstream products of stress kinases and p38 MAPK to promote apoptosis [32]. Among the apoptosisinducing substrates of JNK are Bcl-2 and Bim, which are inhibited and activated, respectively, by JNK phosphorylation [33,34]. In addition, p38 MAPK phosphorylates and activates the transcription factor CHOP, which causes changes in gene expression that favor apoptosis, including increasing expression of Bim and DR5, while decreasing expression of Bcl-2 [15,17, 22,31,35]. The enhancement of JNK and p38 phosphorylation pathways at later time points may be crucial to maintaining the stressed status of the T. vaginalisinfected SiHa cells and promoting apoptosis.
Our data strongly suggest that T. vaginalis infection induced significant ER dysfunction in cervical epithelial cells and activated an autophagic response as a protective mechanism. The brief period in which autophagy seems to be blocked needs further investigation, as it may represent a potential shift in the balance of pro-apoptotic and pro-survival Bcl-2 proteins. In addition, JNK could be a potential regulation point of various signaling pathways involved in the regulation of autophagy and apoptosis in T. vaginalisinfected SiHa cells. Understanding the interaction of ER stress, autophagy, and apoptosis can provide valuable insight into host defense mechanisms and protection against formidable pathogens.
Funding
This work was supported by research fund of Chungnam National University.
Conflicts of Interests
The authors declare that they have no competing interests.
References
- Soper D (2004) Trichomoniasis: under control or undercontrolled? Am J Obstet Gynecol 190(1): 281-290.
- Van Der Pol B, Cynthia Kwok, Bosny Pierre Louis, Anne Rinaldi, Robert A Salata, et al. (2008) Trichomonas vaginalis infection and human immunodeficiency virus acquisition in African women. J Infect Dis 197(4): 548-554.
- Fichorova RN (2009) Impact of T vaginalis infection on innate immune responses and reproductive outcome. J Reprod Immunol 83(1-2): 185-189.
- Mielczarek E, J Blaszkowska (2016) Trichomonas vaginalis: pathogenicity and potential role in human reproductive failure. Infection 44(4): 447-458.
- Tao L, Lili Han, Xia Li, Qi Gao, Lei Pan, et al. (2014) Prevalence and risk factors for cervical neoplasia: a cervical cancer screening program in Beijing. BMC Public Health 14: 1185.
- Ghosh I, Richard Muwonge, Srabani Mittal, Dipanwita Banerjee, Pratip Kundu, et al. (2017) Association between high risk human papillomavirus infection and co-infection with Candida spp. and Trichomonas vaginalis in women with cervical premalignant and malignant lesions. J Clin Virol 87: 43-48.
- Ryan CM, N de Miguel, P J Johnson (2011) Trichomonas vaginalis: current understanding of host-parasite interactions. Essays Biochem 51: 161-175.
- Krieger J N, J I Ravdin, M F Rein (1985) Contact-dependent cytopathogenic mechanisms of Trichomonas vaginalis. Infect Immun 50(3): 778-786.
- Lin W C, Wei Ting Chang, Tsuey Yu Chang, Jyh Wei Shin (2015) The Pathogenesis of Human Cervical Epithelium Cells Induced by Interacting with Trichomonas vaginalis. PLoS One 10(4).
- Fiori PL, P Rappelli, M F Addis, F Mannu, P Cappuccinelli (1997) Contact-dependent disruption of the host cell membrane skeleton induced by Trichomonas vaginalis. Infect Immun 65(12): 5142-5148.
- Lustig G, Christopher M, Ryan, W Evan Secor, Patricia J Johnson (2013) Trichomonas vaginalis contact-dependent cytolysis of epithelial cells. Infect Immun 81(5): 1411-1419.
- Arroyo R, J F Alderete (1989) Trichomonas vaginalis surface proteinase activity is necessary for parasite adherence to epithelial cells. Infect Immun 57(10): 2991-2997.
- Hirt R P, C J Noel, T Sicheritz Ponten, J Tachezy, P L Fiori (2007) Trichomonas vaginalis surface proteins: a view from the genome. Trends Parasitol 23(11): 540-547.
- Stafkova J, Petr Rada, Dionigia Meloni, Vojtěch Žárský, Tamara Smutná, et al. Dynamic secretome of Trichomonas vaginalis: Case study of beta amylases. Mol Cell Proteomics 17(2): 304-320.
- Senft D, Z A Ronai (2015) UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci 40(3): 141-148.
- Urra, H, Estefanie Dufey, Fernanda Lisbona, Diego Rojas Rivera, Claudio Hetz, et al. (2013) When ER stress reaches a dead end. Biochim Biophys Acta 1833(12): 3507-3517.
- Sano R, J C Reed (2013) ER stress induced cell death mechanisms. Biochim Biophys Acta 1833(12): 3460-3470.
- Berridge M J (2002) The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium 32(5-6): 235-249.
- Hetz C (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 13(2): 89-102.
- Walter P, D Ron (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334(6059): 1081-1086.
- Tabas I, D Ron (2011) Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol 13(3): 184-190.
- Woehlbier U, C Hetz (2011) Modulating stress responses by the UPRosome: a matter of life and death. Trends Biochem Sci 36(6): 329-337.
- Shore GC, FR Papa, SA Oakes (2011) Signaling cell death from the endoplasmic reticulum stress response. Curr Opin Cell Biol 23(2): 143-149.
- Rodriguez D, D Rojas Rivera, C Hetz (2011) Integrating stress signals at the endoplasmic reticulum: The BCL-2 protein family rheostat. Biochim Biophys Acta 1813(4): 564-574.
- Quan J H, Byung Hun Kang , Guang Ho Cha, Wei Zhou, Young Bok Koh, et al. (2014) Trichonomas vaginalis metalloproteinase induces apoptosis of SiHa cells through disrupting the Mcl-1/Bim and Bcl-xL/Bim complexes. PLoS One 9(10).
- Kang J H, H O Song, j S Ryu, m H Shin, j M Kim, et al. (2006) Trichomonas vaginalis promotes apoptosis of human neutrophils by activating caspase-3 and reducing Mcl-1 expression. Parasite Immunol 28(9): 439-446.
- Song H O, Myeong Heon Shin, Myoung Hee Ahn, Duk Young Min, Yong Seok Kim, et al. (2008) Trichomonas vaginalis: reactive oxygen species mediates caspase-3 dependent apoptosis of human neutrophils. Exp Parasitol 118(1):59-65.
- Youle R J, A Strasser (2008) The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9(1): 47-59.
- Chang J H, Y S Ryang, S K Kim, J Y Park (2004) Trichomonas vaginalis-induced apoptosis in RAW264.7 cells is regulated through Bcl-xL, but not Bcl-2. Parasite Immunol 26(3): 141-150.
- Galluzzi L, A Diotallevi, M Magnani (2017) Endoplasmic reticulum stress and unfolded protein response in infection by intracellular parasites. Future Sci OA 3(3).
- Puthalakath H, Lorraine A O Reilly, Priscilla Gunn, Lily Lee, Priscilla N Kelly, et al. (2007) ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 129(7): 1337-1349.
- Ron D, S R Hubbard (2008) How IRE1 reacts to ER stress. Cell 132(1): 24-26.
- Lei K, R J Davis (2003) JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc Natl Acad Sci USA 100(5): 2432-2437.
- Deng X, L Xiao, W Lang, F Gao, P Ruvolo, et al. (2001) Novel role for JNK as a stress-activated Bcl2 kinase. J Biol Chem 276(26): 23681-23688.
- Yamaguchi H, H G Wang (2004) CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J Biol Chem 279(44): 45495-45502.
