 Mini Review
Mini ReviewAbstract
As many genes associated with human puberty is also known for their involvement in tumori genesis, WDR11 is also initially recognized as a potential tumor suppressor. The chromosome 10q26 region has been previously contributed to male genital development. In particular WDR11 gene within 10q26 encodes a protein that is a member of the WD repeat protein family to participate in the development and progression of the reproductive system through puberty and adulthood. Recent large analyses add the WDR11 mutations as a potential cause of idiopathic hypogonadotropic hypogonadism (IHH). To date, 14 WDR11 mutations have been shown in IHH patients, 4 of 14 have the second gene mutation and the remaining possess mutation in a single WDR11 gene. The WDR11 protein-signal complex interacts directly with molecules in the development and progression of the reproductive system. We postulate that impaired pubertal development in IHH patients results from a deficiency of proper WDR11 protein interaction in cooperation with additional undetected or known variants.
Keywords: Hypogonadotropic Hypogonadism; Kallmann Syndrome; Delayed Puberty; Gene Mutation; WDR11
Mini Review
The hypothalamic-pituitary-gonadal axis plays animportant role in the development and progression of the reproductive system through puberty and adolescence. This neuroendocrine system is initiated by the decapeptide gonadotropin-releasing hormone (GnRH). The GnRH, secreted by the hypothalamus, stimulates the biosynthesis and the release of gonadotropins from the anterior pituitary gland. These gonadotropins, luteinizing hormone and follicle-stimulating hormone, stimulate the gonads to produce sex steroids and gametes [1-3]. Disruption to the hypothalamic-pituitarygonadal axis can result in hypogonadotropic hypogonadism through deficient production, secretion or action of the gonadotropins [1,4,5]. Idiopathic hypogonadotropic hypogonadism (IHH) may be associated with normosmia or anosmia; co-occurrence of IHH with anosmia is termed Kallmann syndrome. GnRH neurons originate in the olfactory placode/vomeronasal organ region and migrate into the hypothalamus along olfactory neurons. The Kallmann syndrome results from haled migration of GnRH neurons within the meninges and therefore both GnRH and olfactory neurons do not reach the hypothalamus [6-9]. Patients with IHH may also manifest additional anomalies or syndrome such as hearing loss, a variety of neurologic defects, and renal agenesis, midline facial defects, dental agenesis [4,6-9].
More than 30 genes have been implicated in IHH including 9 genes that cause an overlapping syndrome [9-16]. A large degree of variability in inheritance, penetrance and a number of literatures is also seen in IHH and an increasing body of evidence suggests that this disorder can be caused by variants in more than one gene. Variants in known IHH genes currently account for only 50% of IHH cases so more genes are yet to be found. The chromosome 10q26 region has previously been associated with male genital development. WDR11 on chromosome 10 encodes a protein that is a member of the WD repeat protein family and participates in a wide variety of cellular processes. Recent large analyses add the WDR11 mutations as a potential cause of IHH. The WDR11 which accounts for 3 % of IHH mutations [17] is not included in recent large analyses and reviews [18,19]. The purpose of this document is to review the known WDR11 mutations to understand as a potential player in IHH.
WDR11 Gene Mutations
To date, 14 WDR11 mutations have been shown in IHH patients, 4 of 14 have the second gene mutation and the remaining possess mutation in a single, as summarized in Table 1. Figure 1 illustrates the positions of theses variants on WDR11 protein structure. The WDR11 mutations have been associated previously with abnormal male genital development including sperm defects and infertility, hypogenitalism, micropenis, cryptorchidism, small testes (cases 1 to 5, and cases 8 to 11)), and abnormal pubertal development due to hypogonadotropic hypogonadism in female (cases 6 and 7). Digenic mutations, WDR11: c.1303G>A; p.A435Thr and GNRHR: c.275T>C; p.Leu92Pro, are found in one patient (case3 with bilateral cryptorchidism). The variants KAL1: c.1532C>A; p.Ser511Tyr and KAL1: c.190T>C; p.Cys164Arg are detected in case 4 and case 8, respectively, in addition to WDR11 mutation. Case 9 has a micropenis, penoscrotal hypospadias and chordee. The variant, WDR11:c.2409G>T: p.Tyr803Cys, is predicted to exert a pathogenic with a strong conservation. The variant, WDR11:c.1352A>G: p.His451Arg is detected in case 10 with penoscrotal hypospadias and bifid scrotum. Micropenis and cryptorchidism are not observed in this case. Case 11has an atypical micropenis and penile hypospadias, whose variant, WDR11:c.1279T>A: p.Leu427Ileu, may be damaging and highly conserved. Interestingly, in Case 12 with pituitary stalk interruption syndrome (characterized by a triad of thin or interrupted pituitary stalk, an ectopic or defective posterior pituitary gland, and hypoplasia or aplasia of the anterior pituitary gland), two gene mutations, WDR11:c.1306A>G; p.Ileu436Val and PROKR2:c.253C>T; p.Arg85Cys are detected. This observation adds WDR11 mutation as a potential or second cause of the combined pituitary hormonal deficiencies. A more recent study [20] analyzed genetic basis of puberty delayed in both sexes, with all patients exhibiting spontaneous or induced pubertal development before 18 years of age. Of the 59 cases, 15 carried 7 different genes mutations. WDR11 variants are detected in 2 cases (cases 13 and 14 of Table 1).
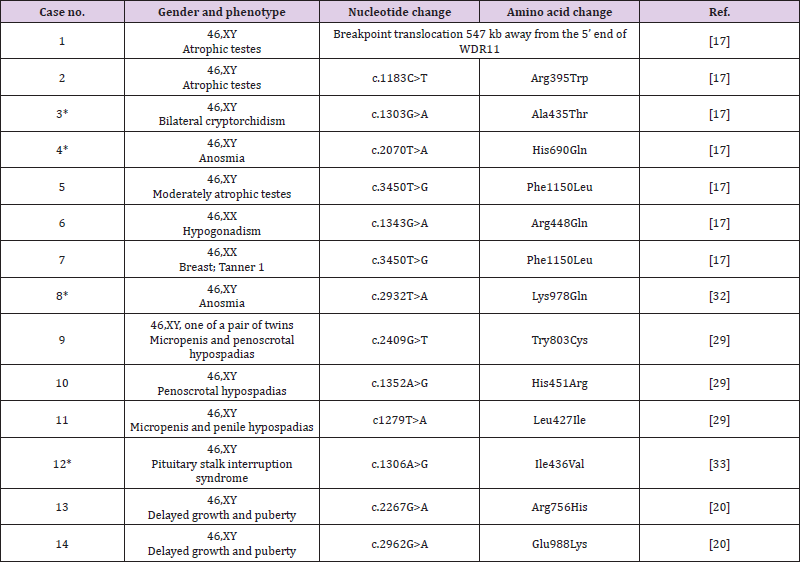
Table 1: Known variants of WDR11 in idiopathic hypogonadotropic hypogonadism patients.
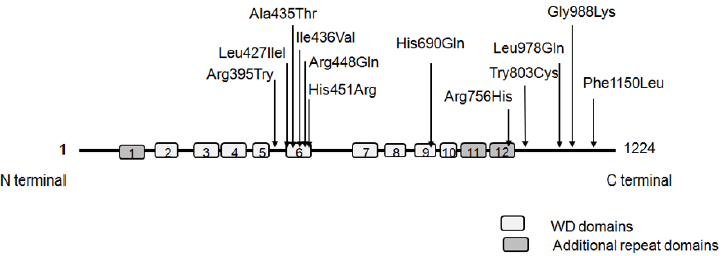
Figure 1: WDR11 protein and the known mutation sites.
WDR11 protein contains 12 WD domains including 3 additional repeat domains. Of the WDR mutations listed in Table 1, 6 directly affect WD domains and others fall within connecting axis.
Discussion
WDR11 was initially recognized as a potential tumor suppressor and its inactivation has been shown to be a part of the multistep process of glial tumorigenesis and tumor progression [21,22]. One of the common frequent genetic changes in glial tumors is heterozygous loss of chromosome that leads to malignant progression. Damages to 10q region have been observed relatively infrequently in low-grade astrocytomas and oligodendrogliomas. Many genes human puberty-linked genes are also known for their involvement in tumorigenesis [23]. For instance, hypothalamic expression of certain tumor suppressor genes is overexpressed during puberty or decreased with delayed puberty [24,25]. The KISS1, a tumor metastasis suppressor gene in melanoma and breast carcinomas encodes a peptide ligand of G protein-coupled receptors, which plays a key role in the initiation of puberty [26,27]. KISS1R mutations cause autosomal recessive IHH in consanguineous families [28]. Although WDR11’s function is unknown, this gene is predicted to display two ß propellers composed of WD domains. WDR11 protein consists of 12 WD domains and nine of them (Figure 1) are involved in the genesis of two consecutive ß propellers [17]. Variants that affect WD domains can disrupt the WD function. The changes predicted to flank the WD domains may also play a significant role in IHH. Of 14 hypogonadism patients with WDR11 variants, 4 patients have mutations in two or more genes. We understand that this sample size is too small, and that future researchis needed to elucidate the pathways involved in the digenic mutations, but these findings serve to suggest which genes could interact with each other. Nevertheless, our analysis may indicate that 10 of 14 patients with hypogonadism patients possess mutation in a single WDR11 gene, indicating that the monogenic mutations account for most cases of IHH and hypogonadism.
Penile and urethral morphology is established before 14 weeks gestation meaning that the fetal hypothalamic-pituitary axis is usually considered to be dispensable for normal penile development, instead relying on placental human chorionic gonadotropin (HCG). After week 14, a continued increase in penile length is dependent upon the hypothalamic-pituitary axis. Thus, boys with hypogonadism often have micropenis but normal phallic morphology [29]. Many individuals have varying degrees of hypospadias [30,31]. Two or more variants contribute to the phenotype in these patients, which may involve additional undetected variants in genes that control either gonadal or penile development. We fail to find common clinical features among mild or reversal variants that would have predicted the clinical course of IHH after adolescence. Of the 14 patients, 6 directly affect WD domains and others fall within connecting axes or additional repeat WD domains (Figure 1). Theses mutations do not always fully penetrate the phenotypic spectrum such as hypogonadism, anosmia, micropenis and penoscrotal hypospadias. Two 46, XX patients (cases 7 and 8) have delayed pubertal development, however, due to young age, it is still unclear whether they enter puberty later in life. A large analysis of 32 reversible IHH patients [18] shows that 2 normosmia male patients carry GNRHR mutation accompanied by another GNRHR mutation and 2 anosmia patients have mutation in CHD7 or FGFR1. We encountered one rare patient with reversal HH after the age of 30 who have mutation WDR11 (manuscript in preparation). Taken together, IHH patients with WDR11 mutations are particularly susceptible to IHH, and phenotypic variability may be inherent to effects of modifier genes and co-occurrence of second gene mutation.
Genetic diagnosis is useful not only for family planning and fertility investigations, but also for direct clinical management. Therapeutic options exist for many of features of IHH. In early life, this may include low-dose testosterone or gonadotropins for micropenis and stimulation of gonadal development. Second, during pubertal or adulthood, testosterone supplement may also induce puberty including psychosocial developments. IHHrelated infertility can also be treated by administering GnRH or gonadotropins [30,31-33]. Therefore, given the treatment options, genetic diagnosis by gene panel testing may allow earlier or bespoke interventions. In summary, to date, 13WDR11 mutations have been shown in 14 IHH patients, 4 of 14 patients have the second gene mutation and the remaining possess mutation in a single WDR11 gene. The WDR11 protein-signal complex interacts directly with molecules on development and progression of the reproductive system. We postulate that impaired pubertal development in IHH patients results from a deficiency of proper WDR11 protein interaction in cooperation with additional undetected or known variants.
Disclosure Statement
The authors declare no conflict of interest regarding the publication of this article.
References
- Boehm U, Bouloux P, Dattani M, De Roux N, Dodé C, et al. (2015) Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism--pathogenesis, diagnosis and treatment. Nat Rev Endocrinol 11(9): 547-564.
- Svechnikov K, Landreh L, Weisser J, Izzo G, Colón E, et al. (2010) Origin, development and regulation of human Leydig cells. Horm Res Paediatr 73(2): 93-101.
- Walker W, Cheng J (2005) FSH and testosterone signaling in Sertoli cells. Reproduction 130(1): 15-28.
- Bhagavath B, Podolsky R, Ozata M, Bolu E, Bick DP, et al. (2006) Clinical and molecular characterization of a large sample of patients with hypogonadotropic hypogonadism. Fertil Steril 85(3): 706-713.
- Schwanzel-Fukuda M, Bick D, Pfaff D (1989) Luteinizing hormone-releasing hormone (LHRH)-expressing cells do not migrate normally in an inherited hypogonadal (Kallmann) syndrome. Mol Brain Res 6(4): 311-326.
- Han T, Bouloux P (2010) What is the optimal therapy for young males with hypogonadotropic hypogonadism? Clin Endocrinol (Oxf) 72(6): 731-737.
- Izumi Y, Suzuki E, Kanzaki S, Yatsuga S, Kinjo S, et al. (2014) Genome-wide copy number analysis and systematic mutation screening in 58 patients with hypogonadotropic hypogonadism. Fertil Steril 102(4): 1130-1136.
- Miraoui H, Dwyer A, Sykiotis G, Plummer L, Chung W, et al. (2013) Mutations in FGF17, IL17RD, DUSP6, SPRY4, and FLRT3 are identified in individuals with congenital hypogonadotropic hypogonadism. Am J Hum Genet 92(5): 725-743.
- Raivio T, Sidis Y, Plummer L, Chen H, Ma J, et al. (2009) Impaired fibroblast growth factor receptor 1 signaling as a cause of normosmic idiopathic hypogonadotropic hypogonadism. J Clin Endocrinol Metab 94(11): 4380-4390.
- Chan Y, De Guillebon A, Lang-Muritano M, Plummer L, Cerrato F, et al. (2009) GnRH1mutations in patients with idiopathic hypogonadotropic hypogonadism. Proc Natl Acad Sci USA 106(28): 11703-11708.
- Cole L, Sidis Y, Zhang C, Quinton R, Plummer L, et al. (2008) Mutations in prokineticin 2 (PROK2) and PROK2 receptor (PROKR2) in human gonadotrophin-releasing hormone deficiency: molecular genetics and clinical spectrum. J Clin Endocrinol Metab 93(9): 3551-3559.
- Dodé C, Teixeira L, Levilliers J, Fouveaut C, Bouchard P, et al. (2006) Kallmann syndrome: mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS Genet 2(10): e175.
- Falardeau J, Chung W, Beenken A, Raivio T, Plummer L, et al. (2008) Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J Clin Invest 118(8): 2822-2831.
- Layman L (2010) In: Weiss RE, Refetoff S, eds. Genetic diagnosis of endocrine disorders. Amsterdam: Academic Press 217-225.
- Pitteloud N, Quinton R, Pearce S, Raivio T, Acierno J, et al. (2007) Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. J Clin Invest. 117(2): 457-463.
- Xu N, Kim H, Bhagavath B, Cho SG, Lee JH, et al. (2011) Nasal embryonic LHRH factor (NELF) mutations in patients with normosmic hypogonadotropic hypogonadism and Kallmann syndrome. Fertil Steril 95(5): 1613-1620.
- Kim H, Ahn J, Kurth I, Ullmann R, Kim HT, et al. (2010) WDR11, a WD protein that interacts with transcription factor EMX1, is mutated in idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet 87(4): 456-479.
- Laitinen E, Tommiska J, Sane T (2012) Reversible congenital hypogonadotropic hypogonadism in patients with CHD7, FGFR1 or GNRHR mutations. PLoS One 7(6): e39450.
- Sykiotis G, Plummer L, Hughes VA, Au M, Durrani S, et al. (2010) Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc Natl Acad Sci USA 107(34): 15140-15144.
- Barroso P, Jorge A, Lerario AM, Montenegro LR, Vasques G, et al. (2019) Clinical and Genetic Characterization of a Constitutional Delay of Growth and Puberty Cohort. Neuroendocrinology Epub ahead of print.
- Chernova O, Hunyadi A, Malaj E, Pan H, Crooks C, et al. (2001) A novel member of the WD-repeat gene family, WDR11, maps to the 10q26 region and is disrupted by a chromosome translocation in human glioblastoma cells. Oncogene 20(38): 5378-5392.
- Smith J, Jenkins R (2000) Genetic alterations in adult diffuse glioma: occurrence, significance, and prognostic implications. Front Biosci 5: D213-D231.
- Ojeda S, Dubay C, Lomniczi A, Kaidar G, Matagne V, et al. (2010) Gene networks and the neuroendocrine regulation of puberty. Mol Cell Endocrinol 324(1-2): 3-11.
- Lee J, Welch D (1997) Suppression of metastasis in human breast carcinoma MDA-MB-435 cells after transfection with the metastasis suppressor gene, KiSS-1. Cancer Res 57(12): 2384-2387.
- Parent A, Matagne V, Westphal M, Heger S, Ojeda S, et al. (2008) Gene expression profiling of hypothalamic hamartomas: A search for genes associated with central precocious puberty. Horm Res 69(2): 114-123.
- Philipps D, Wigglesworth K, Hartford S, Sun F, Pattabiraman S, et al. (2008) The dual bromodomain and WD repeat-containing mouse protein BRWD1 is required for normal spermiogenesis and the oocyte-embryo transition. Dev Biol 317(1): 72-82.
- Ohtaki T, Shintani Y, Honda S, Matsumoto H, Hori A, et al. (2001) Metastasis suppressor gene KiSS-1 encodes peptide ligand of a G-protein-coupled receptor. Nature 411(6837): 613-617.
- Field M, Tarpey P, Smith R, Edkins S, O'Meara S, et al. (2007) Mutations in the BRWD3 gene cause X-linked mental retardation associated with macrocephaly. Am J Hum Genet 81(2): 367-374.
- Ayers K, Bouty A, Robevska G, van den Bergen JA, Juniarto AZ, et al. (2017) Variants in congenital hypogonadotrophic hypogonadism genes identified in an Indonesian cohort of 46,XY under-virilised boys. Hum Genet 11: e1.
- Kurzrock E, Delair S (2006) Hypospadias and Kallmann’s syndrome: distinction between morphogenesis and growth of the male phallus. J Pediatr Urol 2(5): 515-517.
- Vizeneux A, Hilfiger A, Bouligand J, Pouillot M, Brailly-Tabard S, et al. (2013) Congenital hypogonadotropic hypogonadism during childhood: presentation and genetic analyses in 46 boys. PLoS ONE 8: e77827.
- Quaynor S, Kim H, Cappello E, Williams T, Chorich LP, et al. (2011) The prevalence of digenic mutations in patients with normosmic hypogonadotropic hypogonadism and Kallmann syndrome. Fertil Steril 96(6): 1424-1430.
- Mc Cormack S, Li D, Kim YJ, Lee JY, Kim SH, et al. (2017) Digenic Inheritance of PROKR2 and WDR11 Mutations in Pituitary Stalk Interruption Syndrome. J Clin Endocrinol Metab 102(7): 2501-2507.
