 Case Report
Case ReportAbstract
Hereditary Hemochromatosis (HH) is an autosomal recessive disorder due to iron overload that causes severe organ dysfunction. A heterogeneous group of mutations give rise to different HH forms. Hereditary Hemochromatosis type 3, also known as NON-HFE Hemochromatosis (Non-High Iron Hemochromatosis), is a recessive hereditary disease linked to TFR2 gene mutation placed on 7q 22.1 chromosome. HH NON-HFE causes severe iron overload, hepatomegaly, abdominal pain and progressive increased skin pigmentation. Fifteen polymorphisms of HFE (exons 2, 3, 4), TFR2 (exons 2, 4, 6) and FPN1 (exon 5) genes were evaluated in 257 patients (194 male and 63 female). Mutations in HFE, TFR2 and FPN1 genes were evaluated with standard kits. Iron overload was checked out through laboratory testing for serum, ferritin and transferrin saturation. In 257 enrolled patients, a single M172K mutation of TFR2 gene in a 33 years old man has been identified, while no other mutations for HFE and FPN1 genes were observed. Although some HH genetic polymorphisms have been identified, TFR2 gene mutation have not been clearly defined.
Keywords: Hereditary Hemochromatosis Type 3; M172K Mutation; TFR2 gene; Iron Overload
Abbreviations: HH: Hereditary Hemochromatosis; HH type 3: Hereditary Hemochromatosis type 3; TFR2: Transferrin Receptor 2 gene; TFR1: Transferrin Receptor Protein 1; TFR2: Transferrin Receptor Protein 2; FPN1: Ferroportin-1 gene; HJV: Hemojuvelin gene; HAMP: Hepcidin Antimicrobial Peptide gene; SLC40A1: Solute carrier family 40 (iron-regulated transporter) member 1; IRE-IRP: Iron Responsive Element- Iron Dependent Regulatory Protein; EBV: Epstein-Barr Virus; GOT: Glutamate Oxaloacetate Transaminase; GPT: Glutamic- Pyruvic Transaminase; ALP: Alpha-1 Antitrypsin; Gamma- GT: Gamma-Glutamyl Transpeptidase.
Introduction
Hereditary Hemochromatosis (HH) is a genetic disease due to an excessive intestinal dietary iron absorption in basolateral surface of enterocytes. HH represents one of the most common genetic disease in North Europe. This suggests that genetic mutation leading to hemochromatosis originally occurred in Celtic populations [1]. In 1865, Trousseau described a new syndrome characterized by diabetes, pigmented liver, cirrhosis and bronze-colored skin.In 1889, Von Recklinghausen coined the term hemochromatosis to connect the symptoms reported, 24 years earlier, by Trousseau to an abnormal iron deposition in tissues and organs. In 1935, Sheldon classed this disease as an inborn error of metabolism [2]. According to Online Mendelian Inheritance in Man (OMIM), 5 types of HH have been identified and classified on the basis of different clinical, biochemical, and genetic characteristics [3]:
1) HH type 1 is due to HFE gene mutations which is linked on chromosome 6. HH type 1 is also known as Classic Hereditary Hemochromatosis and represents the most common genetic iron overload disorder in Caucasian populations [2]. HFE gene encodes for membrane protein which is similar to major histocompatibility class I-like (MHC1). Mutation in HFE gene generates a protein unable to interact with TFR1 protein (transferrin receptor 1). This affects iron overload in liver, heart, pancreas and skin leading to liver damage, myocarditis, diabetes and hypogonadism. The majority of HH type 1 cases (approximately 80%) is homozygous for a mutation linked to the substitution of a tyrosine residue for cysteine at position 282 (C282Y) [3].
2) HH type 2 (juvenile) is considered the most severe form of rare HH. First symptoms of iron overload typically begin during childhood and include low sex-hormones level and heart disease. HH type 2 is divided into two forms: HH type 2A caused by mutation in HJV gene linked on chromosome1; HH type 2B due to HAMP gene mutation linked on chromosome 19. Serum and urinary hepcidin levels are decreased in both mutations. Juvenile hemochromatosis symptoms are like those seen in HH type 1 and occur at an early age and are usually more severe [4]. 3) HH type 3 is caused by mutation in TFR2 gene linked on long arm of human chromosome 7 (7q22.1). Patients with HH type 3 show first signs and symptoms (fatigue, hepatomegaly, abdominal pain, arthralgia, progressive increase in skin pigmentation) of iron overload in the third decade. TFR2 gene encodes for homologue protein of transferrin receptor 1 (TFR1) [2].
4) HH type 4 is caused by mutations in Ferroportin encoding gene (SLC40A1) linked on chromosome 2. This differs to other forms for its autosomal dominant inheritance. HH type 4 can be further divided into two subtypes: type 4A includes patients with first liver disease in oldness; type 4B (associated with fatigue, weakness, joint pain, abdominal pain, loss of libido, liver disease, diabetes, heart problems, difficulty breathing and skin discoloration) can begin anytime from childhood to adulthood [2] (Table 1).
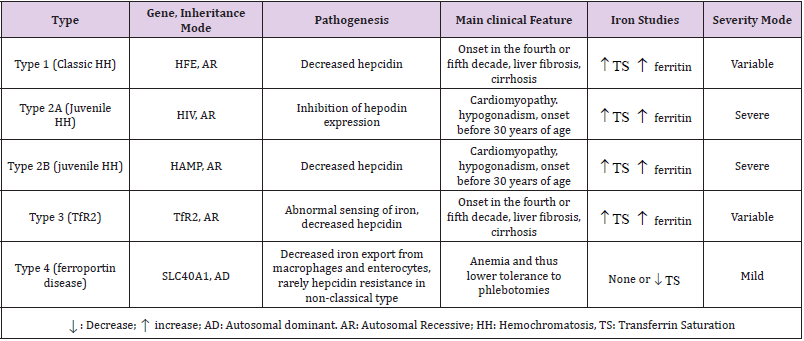
Table 1: Types of hereditary hemochromatosis, genes mutation and symptoms [5].
Table abbreviations: HH: Hereditary Hemochromatosis; HFE: High Fe gene; HJV: Hemojuvelin gene; HAMP: hepcidin antimicrobial peptide gene; TFR2: Transferrin Receptor 2 gene; SLC40A1: solute carrier family 40 (iron- regulated transporter), member 1; AD: Autosomal dominant; AR: Autosomal recessive; TS: Transferrin saturation.
HH type 3 is considered a rare recessive disorder and single families with TFR2 gene mutation have been identified in France, Portugal, Spain and Taiwan [5,6]. TFR2 gene encodes for homologue protein of transferrin receptor 1 (TFR1) [2]. Nucleotide and amino-acid sequence of TFR1 and TFR2 are homologous although different tissue expression has been reported. TFR1 is ubiquitously expressed, whereas the expression of TFR2 is restricted to the hepatocytes and erythroid precursors [6]. TFR2 is a 21 Kb long gene with 18 exons subjects to alternative splicing. This gene encodes for two variants: TFR2α and TFR2β. TFR2 transcription is unresponsive to intracellular iron levels unlike TFR1, as demonstrated by the absence of IRE-IRP (iron responsive element - iron dependent regulatory protein) elements in TFR2 mRNA [7]. IRPs can bind to RNA stem-loops containing an IRE in the 3’-5’ UTR to manipulate translation of target mRNA in accordance with changes of intracellular iron level. In condition of iron deficiency, the IRP binds IRE in the 5’UTR of Ferritin mRNA and inhibits translation, whereas binding to the IRE present in 3’- UTR of TFR1 mRNA prevents nuclease degradation. This increases iron uptake and prevents iron storage. In condition of iron excess, the IRP does not inhibit Ferritin expression while TFR1 is clavated by endonucleases and excess of iron can be stored adequately [8]. Transferrin receptor 2 function is still unclear. Recent studies suggested that TFR2 plays an important role in the iron-dependent regulation of peptide hepcidin, a key regulator of body iron homeostasis expressed in liver [2]. Furthermore, mutant mice for TFR2 gene showed hepcidin down-regulation. This suggest that TFR2 is an essential gene in the pathway of iron homeostasis [9]. Hepcidin is synthesized predominantly in hepatocytes and is considered the principal regulator of iron absorption. Dysregulation of hepcidin production results in a variety of iron disorders. Hepcidin deficiency is associated with systemic iron overload and Hereditary Hemochromatosis [8].
5) Genetic sequencing of TFR2 gene revealed 5 mutational sequences:
a) A C→G missense mutation in exon 6 of TFR2 gene that determines an amino-acid change in the protein, from Tyrosine (UAC) to a premature stop codon (UAG). This is known as Y250X mutation and causes inactivation of both TFR2 transcripts (TFR2α-TFR2β) [10].
b) A poly-C site expansion (84-88 insC) in exon 2 that causes a frameshift and the transcription of premature stop codon on 60 amino-acid. This mutation E60X inactivates TFR2α transcript [10].
c) A 12 base pair deletion in exon 16 that causes the loss of 4 amino acid (AVAQ) on TfR2 extracellular protein domain [10].
d) A missense mutation on exon 17 (Q690P) [10].
e) A T→A transversion (T515A), which causes a Methionine→Lysine
f) Substitution at 172 position (M172K). The replacement from basic to neutral amino acid reduces TFR2 protein expression on the surface of the cell membrane [10].
M172K mutation is considered one of the rarest sequence variants of TFR2 gene. This transversion causes a missense mutation in α transcript which can prevent the β form transcription [11]. The lack of β transcript is associated with severe phenotype for M172K homozygosity. Patients with M172K show low levels of hepcidin, due to a dysfunction in transduction pathway of TFR1- HFE-TFR2 complex [11] (Figure 1).
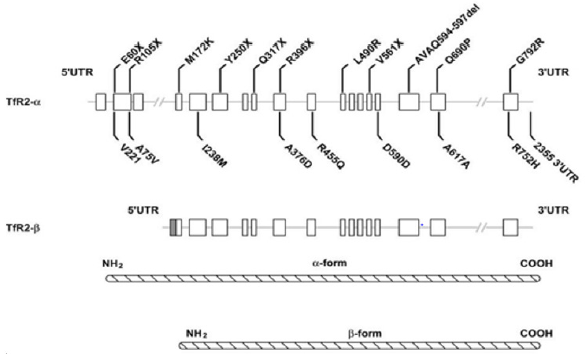
Figure 1: Schematic representation of TFR2 mutations. Two transcripts, possibly resulting from alternative splicing (as suggested by Kawabata et al. (1999)), are shown. Compared to TFR2-α transcript, which contains 18 exons (whiteboxes), the β-transcriptlacksexons 1-3 and hasanadditional 142 nucleotidesatthe5’-end(greybox).The causal mutations are reported above the gene sequence, while the polymorphic changes are illustrated below the gene. TFR2 α-protein and β-protein are also shown[12].
Case Report
At 31 July 2018 a 33-old male with normal medical history, was hospitalized with a suspected hepatitis in Gastroenterology Department at “Santa Maria” Hospital in Terni. Blood analysis revealed critical values:Hepatic or splenic injury was confirmed by high levels of transaminases. Immunological tests were carried out to exclude viral hepatitis as a cause of high levels of transaminases. Unexpectedly, anti-EBV antibody levels were high. These results confirmed hepatitis due to Epstein–Barr virus. Elevated values of plasmatic Ferritin can address the suspect to an iron overload disorder. “DNA extraction” and “Hemochromatosis 15 mutations” standard kits were used to reveal mutations in HFE, TFR2 and FPN1 genes. This kit identifies 15 polymorphisms of HFE (exons 2, 3, 4); TFR2 (exons 2, 4, 6) and FPN1 (exon 5) by DNA isolation from whole blood (Tables 2 & 3).
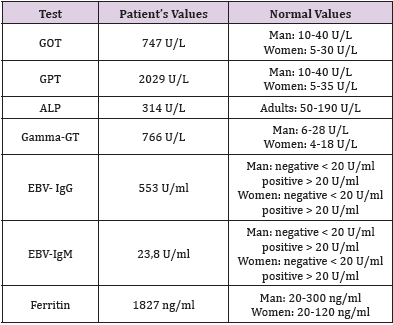
Table 2: Significant abnormal values in patient with M172K mutation hospitalized for suspected hepatitis.
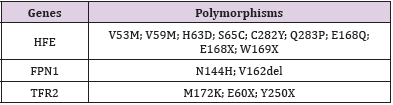
Table 3: Representation of principal polymorphism identified by “Hemochromatosis 15 polymorphism” standard kit.
Results
In 257 enrolled patients, this test revealed a single M172K mutation only in a 33 years old man; no other mutations for HFE and FPN1 genes, were observed in the same case.
Discussion
HH is the principal cause of systemic iron overload. It is known that HH can be due to different mutations in different genes. The lack of knowledge and the absence of diagnostic tools make the diagnosis very complex. In this study, the molecular analysis prompts to formulate an early diagnosis of TFR2-related (type 3) HH that increases the chances for successful treatment. This work adds more information about HH type 3 suggesting that M172K mutation could be more frequently involved in systemic primary iron overload than has been thought.
References
- Distante S, Robson KJH, Graham-Campbell J, Arnaiz Villena A, Brissot P, et al. (2004) “The origin and spread of the HFE-C282Y haemochromatosis mutation”. Hum Genet 115(4): 269-279.
- Le Gac G,Férec C (2005) “The molecular genetics of haemochromatosis”. Eur JHum Genet 13(11): 1172-1185.
- Santos PCJL, Krieger JE, Pereira AC (2012) “Molecular diagnostic and pathogenesis of hereditary hemochromatosis”. Int J Mol Sci 13(2): 1497-1511.
- Babitt JL, Lin HY (2011) “The molecular pathogenesis of hereditary hemochromatosis”.Semin Liver Dis 31(3): 280-292.
- Kanwar P, Kowdley KV (2013) “Diagnosis and treatment of hereditary hemochromatosis: An update”. Expert Rev. Gastroenterol Hepatol 7(6): 517-530.
- Silvestri L, Nai A, Pagani A, Camaschella C (2014) “The extrahepatic role of TFR2 in iron homeostasis”. Front Pharmacol 5: 1-7.
- Lee PL, Halloran C, West C,Beutler E (2001) “Mutation analysis of the transferrin receptor-2 gene in patients with iron overload”. Blood Cells Mol Dis 27(1): 285-289.
- Pantopoulos K (2004) “Iron Metabolismo and the IRE/IRP Regulation System”. Ann N YAcad Sci 1012: 1-13.
- Drake SF (2007) “Iron absorption and hepatic iron uptake are increased in a transferrin receptor 2 (Y245X) mutant mouse model of hemochromatosis type 3”. Am J Physiol - Gastrointest. Liver Physiol 292(1): 323-328.
- Roetto A (2002) “Hemochromatosis due to mutations in transferrin receptor 2”. Blood Cells Mol Dis 29(3): 465-470.
- Roetto A, Garozzo G,Calì, M De Gobbi, Gasparini P, et al. (2001) “New mutations inactivating transferrin receptor 2 in hemochromatosis type 3,” Blood 97(9): 2555-2560.
- Calzolari A, Oliviero I, Testa U (2007) “Transferrin receptor 2 is emerging as a major player in the control of iron metabolism.” Cent Eur J Biol 2(1): 34-55.
