 Research Article
Research ArticleAbstract
Prostate cancer is a debilitating disease that kills thousands of men worldwide each year. Investigating the biology of prostate cancer remains a high priority on the global scientific landscape. The current study utilized microarray techniques (Affymetrix Human Genome U133 Plus 2.0 Arrays) and bioinformatics analysis to compare the gene expression profiles of human primary prostate epithelial cells (HPrEC) and DU145 human prostate cancer cells. Microarray analysis yielded 6310 differentially expressed genes (DEGs) with a two-fold change and 1322 DEGs with a five-fold change. Metascape was used to characterize differentially expressed genes to specific molecular networks and canonical signaling pathways. Several signal transduction pathways and relevant prostate cancer-related genes (e.g., CSF1, CTSL, CYP3A5, DPYSL3, EGFR, FYN, LAMB3, ITGB4, PYGB) were identified which are consistent with previous studies as potential targets to prostate cancer progression. Future research publications using the current microarray dataset will explore the biological functions of select differentially expressed genes and protein products to better resolve the molecular basis of prostate cancer development and progression and may elucidate biomarkers and novel therapeutic strategies designed to effectively target prostate cancer.
Keywords: Gene Expression; Bioinformatics; Microarray; VEGF Signaling Pathway; Prostate Cancer
Abbreviations: HPrEC: Human Primary Prostate Epithelial Cells; DEGs: Differentially Expressed Genes; PSA: Prostate Specific Antigen; DMEM: Dulbecco’s Modified Eagle Medium; RMA: Robust Multi-Chip Analysis; MCODE: Molecular Complex Detection; DRE: Digital Rectal Examination; KEGG: Kyoto Encyclopedia of Genes and Genomes
Introduction
Prostate cancer remains the second leading cause of cancerassociated deaths of men in the United States [1]. The prostate gland is a component of the male reproductive system in mammals roughly the size of a walnut. The prostate gland is below the urinary bladder and is responsible for the secretion of an alkaline fluid that provides nourishment and protection for spermatozoa. Prostate cancer is the result of genetic and epigenetic factors that lead to the unregulated growth of prostate cells [2]. The use of the Gleason score, prostate specific antigen (PSA) levels, and the TNM system are often used to characterize prostate cancer and to determine the appropriate medical response. Disease surveillance data suggests that incidence and mortality of prostate cancer overwhelmingly occurs in men over 50 years old with family history as a major predisposition factor.Reports also suggest that early detection significantly leads to higher survival rates. Prostate cancer treatments in recent years include a combination of hormone therapy, surgical procedures, and chemotherapy [3]. The elucidation of underlying molecular mechanisms of prostate cancer can improve the detection, diagnosis, and disease staging. The identification of potential biomarkers and therapeutic solutions may lead to the reduction of global prostate cancer cases and deaths.
The development of biomolecular microarrays over 30 years ago has exponentially improved our comprehension of the molecular events that underlie biological processes in prokaryotes and eukaryotes [4]. The utilization of DNA microarrays and bioinformatics to evaluate gene expression of human carcinomas is a widely used technique to gain insights into disease development, progression, biomarkers, diagnosis, and prognosis [5-8]. One of the more exciting applications of microarrays is to utilize DNA chips to explore gene expression profiles specific to various therapeutic treatments [9,10]. Ren, et al. [9] used microarrays to test the potential therapeutic effects of Salvia miltiorrhiza to treat cardiovascular diseases. The researchers treated different components of Salvia miltiorrhiza (e.g., hydrophilic and lipophilic) as well as complete extracts on human umbilical vein endothelial cells and analyzed the underlying molecular response via gene expression profiling. Data revealed that the lipophilic component of Salvia miltiorrhiza, not the hydrophilic component, upregulated genes that may serve important functions in ameliorating specific cardiovascular diseases. Drug repurpose evaluations can also be conducted using microarray technology.
Recently, researchers identified the DEGs involved in adipogenesis and used the drug screening approach (connectivity map) to determine that the drug Pyrvinium, an existing antihelmintic drug, would serve as a potent anti-adipogenic agent based on inverse DEGs [10]. The purpose of the current study was to identify specific molecular components and biological pathways associated with prostate cancer using microarray technology and bioinformatics analysis. Metascape was employed to analyze gene expression profiles of DU145 cells and HPrECs. The research question guiding this study was: What are the differences in gene expression patterns between normal and cancerous prostate cells? Insight into specific upregulated or downregulated genes and overactive or nonactive biologic pathways may provide acute understanding of prostate cancer treatment strategies as discussed in previous gene expression prostate cancer studies [11-13]. This study offers a seminal glance at comparing the gene expression profiles of DU145 cells and primary prostate epithelial cells. The synergy of microarray studies and bioinformatics analysis will positively affect research investigations into the underlying molecular causes of human cancers.
Materials and Methods
Prostate Cell Culture
Human primary prostate epithelial cells (HPrEC), DU145 human prostate cancer cells, and cell culture reagents were purchased from the American Type Culture Collection (Manassas, Virginia). Primary prostate epithelial cells were cultured in prostate epithelial cell basal media supplemented with a prostate epithelial cell growth kit and penicillin-streptomycin solution (ATCC). DU145 prostate cancer cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% FBS and penicillin-streptomycin solution. Human cells were cultured for 72 hrs. at 37°C in a 5% CO2 atmosphere and harvested as previously described [14]. Prostate cells were analyzed using an Olympus IX70 Microscope to determine cell morphology and stringency of culture conditions.
RNA Sample Preparation and Microarray Procedures
Total RNA was extracted from HPrECs and DU145 cells using the RNeasy Plus Mini kit (Qiagen, Gaithersburg, MD) according to the manufacturer’s specifications and stored at -70°C. HPrEC and DU145 RNA samples were submitted to the Duke University Microarray Facility (Durham, NC) for processing. RNA quantity and quality were determined using the Agilent 2100 Bioanalyzer System. Affymetrix Human Genome U133 Plus 2.0 Arrays were used. Robust multi-chip analysis (RMA) normalization procedures were performed prior to bioinformatics software utilization. ANOVA and fold-change were used to detect differentially expressed genes.
Analysis of the microarray dataset was performed at the Duke University Microarray Facility using Partek Genomics Suite statistical analysis software. The Metascape [15] bioinformatics software program was used in the Microbial Signal Transduction Pathway Research Lab at Livingstone College. Metascape (http:// metascape.org) computational procedures allowed for an in-depth analysis of signal transduction pathways and molecular networks associated with human prostate cancer cells. Figure 1 shows the workflow of the overall experiment (Figure 1).
Figure 1: Schematic of the experimental protocol.
Results
Differentially Expressed Genes
Following analysis of RMA normalized microarray data, it was determined that a total of 6310 DEGs with a two-fold change or greater (p-value of <0.05) and 1322 DEGs with a five-fold change or greater (p-value of <0.01) were generated (Figure 2). The gene expression profile of HPrECs provided the baseline in this experiment.

Figure 2: Differentially expressed genes in DU145 cells compared to HPrECs (baseline).
Gene Ontology Enrichment Analysis of DEGs
In the remainder of the Results section only the 1322 DEGs that demonstrated a five-fold change or greater were analyzed. Figure 3 illustrates a heat map based on gene ontology analysis that displays the major biological processes associated with the selected DEGs. Only the top 20 clusters are shown. Bar color intensity reflects statistically significant p-values. Further, analysis indicates that the majority of the genes were involved in epidermis development, extracellular matrix organization, and tissue morphogenesis. The top 20 gene clusters illustrated in the heat map are extrapolated in Table 1 to show the number of genes in each cluster and the percentage of the genes found associated with each gene ontology term (Figure 3 & Table 1).
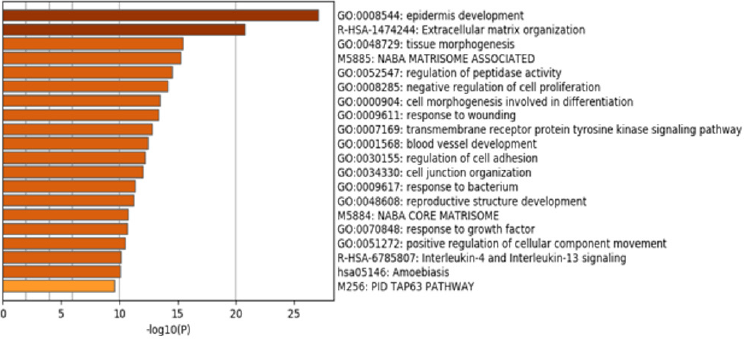
Figure 3: Heat map derived from GO enrichment analysis of DEGs with a five-fold change or greater.
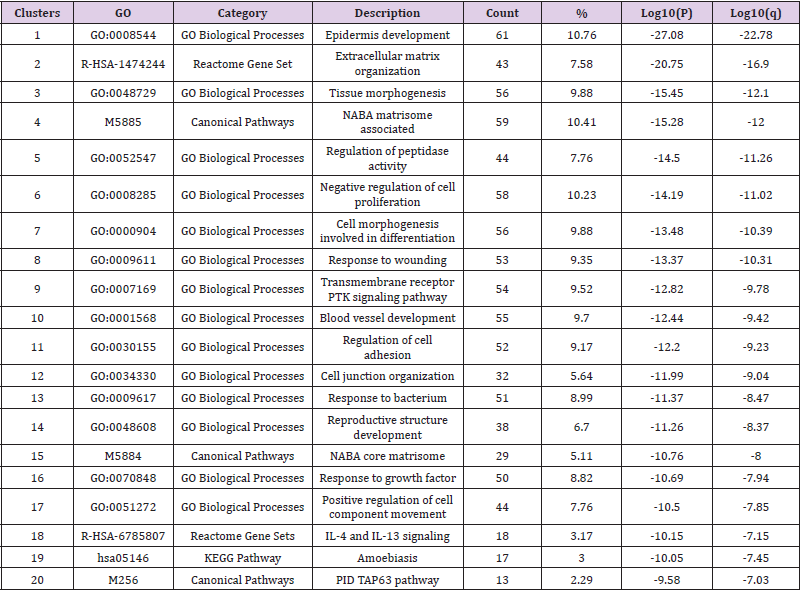
Table 1: Biological signal pathway and biological process enrichment evaluation.
Protein-Protein Interaction Analysis and MCODE Analysis
Figure 4 illustrates the protein-protein network. Proteins in the network are believed to interact with each other to promote specific biologic processes on the basis of experimental and computational data. The protein-protein interaction network was analyzed using molecular complex detection (MCODE) module analysis as previously performed [16,17]. MCODE procedures further subdivided the initial protein-protein interaction map into 8 subgroups differentiated by color and number: MCODE 1 - red, MCODE 2 - blue, MCODE 3 - green, MCODE 4 - purple, MCODE 5 - orange, MCODE 6 - yellow, MCODE 7 - brown, MCODE 8 - pink. Based on the module analysis shown in Figure 5 several genes were identified: CSF1, CTSL, CYP3A5, DPYSL3, EGFR, FYN, LAMB3, ITGB4, and PYGB. Related biological processes identified by MCODE analysis consisted of cell cycle, antigen processing and presentation, VEGF signal transduction pathway, cell adhesion, platelet activation, Ras signaling transduction pathway, and PI3KAkt signaling pathway (Figures 4 & 5).
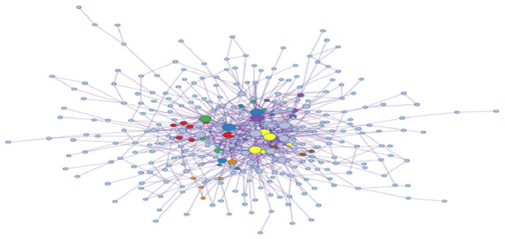
Figure 4: Protein-protein network analysis.
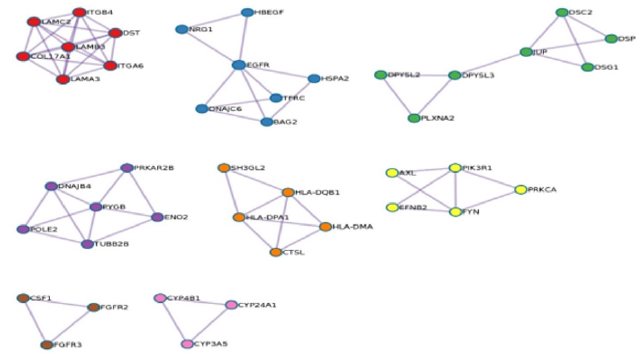
Figure 5: MCODE protein-protein network.
Discussion
Prostate cancer will account for the highest number of new cancer cases in men in the United States according to a recent report [1]. Dysregulated signaling mechanisms have been implicated in negative pathophysiological outcomes associated with prostate cancer. Screening methods such as prostate specific antigen (PSA) and the lesser utilized digital rectal examination (DRE) are still the main prostate cancer early detection methods. It is generally known that higher than normal PSA levels in men suggests potential prostate pathology. It should be noted that evaluation of PSA levels in isolation can be misleading given the current understanding that PSA levels can be affected by a number of factors that are not related to prostate cancer such as age, race, and microbial infections. While biochemical analysis and physical inspection are essential early detection techniques in the fight against diagnosing prostate cancer, the elucidation of diagnosis and prognosis determination at the molecular level remains a dedicated focus of prostate cancer biologists [18].
Comparative gene expression profiling is a useful high throughput tool to identify differences in cells, tissues, and disease mechanisms [19-22]. Microarray procedures coupled with bioinformatics analysis of gene expression profiles of normal and abnormal tissues can lead to the discovery of target signal transduction pathways and molecules that may play a role in aberrant cellular differentiation and disease [23]. In the current study, the gene expression activity of DU145 human prostate cancer cells and human primary prostate epithelial cells (HPrEC) was compared using microarray technology and bioinformatics procedures utilizing the Metascape and Kyoto Encyclopedia of Genes and Genomes (KEGG) platforms. Bioinformatics analysis revealed a total of 6310 differentially expressed genes (2-fold difference, p<=0.05) and a total of 1322 differentially expressed genes (5-fold difference, p<=0.01). The data also revealed a number of potentially promising biomarkers and molecular candidates for which therapeutic intervention may yield positive results.
The protein-protein interaction map (Figure 4) provides an illustration of the transient and stable protein associations that play a role in DU145 prostate cancer cells. While some or many of the protein-protein interactions displayed in Figure 4 may not be essential in maintaining tumor survival, disrupting the dynamics of critical protein relationships may represent a viable therapeutic strategy to treat prostate cancer. MCODE analysis revealed that cell cycle mechanisms, Rap1 signaling pathway, Ras signaling pathway, PI3K-Akt signaling pathway, and VEGF signaling are particularly relevant. The diversity of the MCODE subgroups displayed in Figure 5 is not surprising given the understanding that human tumorigenic processes are complicated and involve a large number of molecular interactions. The VEGF signaling pathway has been implicated in pro-tumorigenic processes such as angiogenesis and metastasis [24,25]. Binding of the VEGF to the vascular endothelial growth factor receptor results in gene expression that promotes the production of blood vessels, cell migration, and cell survival.
The development of new blood vessels is necessary for tumor development and a major requirement for tumor invasivity. Figure 6 was obtained from KEGG and displays the VEGF canonical signal transduction pathway [26]. Targeting the VEGF signaling pathway has been successfully applied to human cancers such as breast cancer, non-small cell lung cancer, and prostate cancer [27,28]. Specifically, Bevacizumab, a monoclonal antibody that targets VEGF along with docetaxel inhibited cell proliferation processes in prostate cancer [29]. Proteins identified in this study could be used as therapeutic targets to suppress cellular growth. Figure 6 displays additional potential downstream targets of VEGF signaling that may produce anti-tumor effects on prostate cancer cells (Figure 6).This seminal study was designed to present global gene expression data to identify specific biomarkers and therapeutic targets. These types of studies are important and have the potential to convey meaningful insights regarding neoplastic transformations.
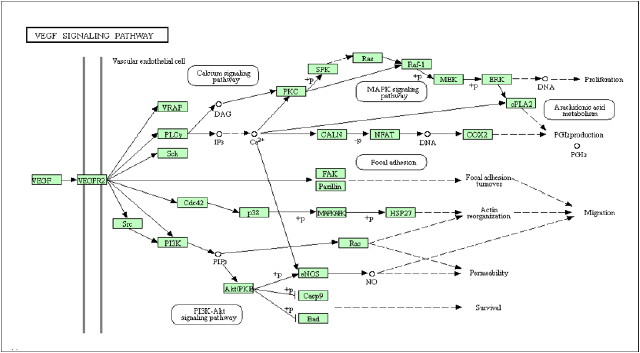
Figure 6: VEGF-A associated with brain edema.
Cell culture-based gene expression profile studies, however, should be followed with microarray procedures involving primary tissue from patients to more accurately evaluate endogenous biological conditions. Comparing the molecular profiles (e.g., gene, protein) of healthy and diseased tissue from patients will lead to more robust data regarding the innerworkings of malignant neoplastic diseases. Future research studies in the Microbial Signal Transduction Pathway Research Lab at Livingstone College will focus on select DEGs that were highly upregulated or downregulated in DU145 cells compared to human primary prostate epithelial cells. Additionally, subsequent investigations will involve the treatment of DU145 cells with existing drugs in a dose-dependent manner coupled with gene expression profiling (e.g., microarray and PCR array). These studies will be designed to evaluate the therapeutic value of synthetic and natural drugs in treating prostate cancer and may implicate new target molecules and biological processes of interest and expand our understanding of the development and progression of prostate cancer.
Acknowledgement
The author would like to thank the Duke University Microarray Facility for their support in performing the microarray experiments. This work was supported by a grant funded by the National Science Foundation (HRD-1505098).
References
- Siegel R, Miller K, Jemal A (2020) Cancer statistics, 2020. CA: A cancer journal for clinicians 70(1): 7-30.
- Cooper C, Foster C (2009) Concepts of epigenetics in prostate cancer development. British Journal of Cancer 100(2): 240-245.
- Teo M, Rathkopf D, Kantoff P (2019) Treatment of advanced prostate cancer. Annual Review of Medicine 70: 479-499.
- Fodor S, Rava R, Huang X, Pease A, Holmes C, et al. (1993) Multiplexed biochemical assays with biological chips. Nature 364(6437): 555-556.
- Macgregor P (2003) Gene expression in cancer: The application of microarrays. Expert Review of Molecular Diagnostics 3(2): 185-200.
- Musso R, Pucci G, Spada M, Fazio I, Russo G, et al. (2019) Gene expression profiles induced by high-dose ionizing radiation in MDA-MB-231 triple-negative breast cancer cell line. Cancer Genomics & Proteomics 16(4): 257-266.
- Shangguan H, Tan S, Zhang J (2015) Bioinformatics analysis of gene expression profiles in hepatocellular carcinoma. European Review for Medical and Pharmacological Sciences 19(11): 2054-2061.
- Tao Z, Shi A, Li R, Wang Y, Wang X, et al. (2017) Microarray bioinformatics in cancer-A review. Journal of BUON 22(4): 838-843.
- Ren G, Fan X, Liang Q, Wang Y, Luo G (2013) Screening and evaluation of traditional Chinese medicine by microarray expression analysis. Journal of Ethnopharmacology 147(3): 564-569.
- Wang Z, Dai Z, Luo Z, Zuo C (2019) Identification of pyrvinium, an anthelmintic drug, as a novel anti-adipogenic compound based on the gene expression microarray and connectivity map. Molecules 24(13): 1-13.
- Chandran U, Dhir R, Ma C, Michalopoulos G, Becich, M, et al. (2005) Differences in gene expression in prostate cancer, normal appearing prostate tissue adjacent to cancer and prostate tissue from cancer free organ donors. BMC Cancer 5: 45.
- Chen W, Wang G (2012) Gene expression profiling of cancer stem cells in the DU145 prostate cancer cell line. Oncology Letters 3(4): 791-796.
- Rapin N, Bagger F, Jendholm J, Mora Jensen H, Krogh A, et al. (2014) Comparing cancer vs normal gene expression profiles identifies new disease entities and common transcriptional programs in AML patients. Blood 123(6): 894-904.
- Flowers L, Subramaniam P, Johnson H (2005) A SOCS-1 peptide mimetic inhibits both constitutive and IL-6 induced activation of STAT3 in prostate cancer cells. Oncogene 24(12): 2114-2120.
- Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi A, et al. (2019) Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nature Communications 10(1): 1523.
- Bader G, Hogue C (2003) An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 4: 2.
- Liu J, Zhou S, Li S, Jiang Y, Wan Y, et al. (2019) Eleven genes associated with progression and prognosis of endometrial cancer (EC) identified by comprehensive bioinformatics analysis. Cancer Cell International 19: 136.
- Shtivelman E, Beer T, Evans C (2014) Molecular pathways and targets in prostate cancer. Oncotarget 5(17): 7217-7259.
- Fang E, Zhang X, Wang Q, Wang D (2017) Identification of prostate cancer hub genes and therapeutic agents using bioinformatics approach. Cancer Biomarkers 20(4): 553-561.
- Flowers L (2019) Bioinformatics analysis to explore ischemic stroke. International Journal of Biology, Pharmacy and Allied Sciences 8(7): 1290-1299.
- Moody L, Herbst A, Yoo H, Vanderloo J, Aiken J (2009) Comparative prion disease gene expression profiling using the prion disease mimetic, cuprizone. Prion 3(2): 99-109.
- Zhang Q, Chen W, Chen S, Li S, Wei D, et al. (2019) Identification of key genes and upstream regulators in ischemic stroke. Brain and Behavior 9(7): e01319.
- Hamilton S, White E, Washington E, Flowers L (2016) Utilization of microarray analysis to determine therapeutic targets in human cancers. International Journal of Biosciences 8(2): 95-105.
- Bender R, Mac Gabhann F (2015) Dysregulation of the vascular endothelial growth factor and semaphorin ligand-receptor families in prostate cancer metastasis. BMC Systems Biology 9: 55.
- Washington E, Flowers L (2018) Analysis of regulatory molecules that mediate metastasis and angiogenesis in human cancer. International Journal of Biology, Pharmacy and Allied Sciences 7(5): 912-922.
- Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M (2004) The KEGG resource for deciphering the genome. Nucleic Acids Research 32: D277-280.
- Ellis L, Hicklin D (2008) VEGF-targeted therapy: Mechanisms of anti-tumour activity. Nature Reviews Cancer 8(8): 579-591.
- Melegh Z, Oltean S (2019) Targeting angiogenesis in prostate cancer. International Journal of Molecular Sciences 20(11): 2676.
- Ortholan C, Durivault J, Hannoun Levi J, Guyot M, Bourcier C, et al. (2010) Bevacizumab/docetaxel association is more efficient than docetaxel alone in reducing breast and prostate cancer cell growth: A new paradigm for understanding the therapeutic effect of combined treatment. European Journal of Cancer 46(16): 3022-3036.
