 Research Article
Research ArticleAbstract
A group of 15 patients (13 with classic phenylketonuria (PKU) under diet therapy and 2 with mild PKU under free diet and pharmacological therapy with sapropterin dihydrochloride (Kuvan®)), was evaluated in just over one year retrospective analysis with the aim to assess, for each patient, at least for three times, plasmatic concentrations of phenylalanine (Phe) and Phenylalanine/Tyrosine (Phe/Tyr) ratio, using Liquid Chromatography Electrospray Ionization Tandem Mass (LC-ESI-MS/ MS). All patients undergone early diagnosis through neonatal screening and therefore all of them were timely treated. No patient developed neurocognitive delay. We propose the use of Phe/Tyr ratio, more than classic evaluation of Phe value only, as prognostic metabolic biomarker of good or poor metabolic control or rather the ability to convert phenylalanine into tyrosine, reducing accumulation during patient followup. Moreover, although phenylalanine is the primary marker for assessing correct adherence dietary and pharmacological treatments, these preliminary data suggest that Phe/Tyr ratio could give a more real idea of diet adherence and pharmacological treatments in every patient. In fact, a good Phe/Tyr ratio should be expression of a better diet compliance in classic PKU patients, and of a good tyrosine amount both from conversion of phenylalanine and from free diet in patients affected by mild PKU, undergo Kuvan® therapy. A prospective analysis, with a longtime patients follow-up and a population study more representative, could confirm this possible role.
Keywords:Phenylketonuria; Hyperphenylalaninemia; Metabolic Control; Therapy Adherence; Newborn Screening; Mass Spectrometry
Abbreviations:PKU: Phenylketonuria; HPA: Hyperphenylalaninemia; Phe: Phenylalanine; Tyr: Tyrosine; Phe/Tyr: Phenilalanine/ Tyrosine; PAH: Phenylalanine Hydroxylase; BH4: Tetrahydrobiopterin, LC-ESI- MS/MS: Liquid Chromatography Electrospray Ionization Tandem Mass; MRM: Multiple Reaction Monitoring; DBS: Dried Blood Spot; PHE: Plasmatic Concentration of Phenylalanine
Introduction
Expanded newborn screening has radically changed natural history and therapeutic management of many pediatric metabolic diseases. Early diagnosis permits to start very precociously diet or pharmacological therapies. This new approach allows to taper the development of the clinical phenotype of these diseases, avoiding, or in any case limiting, the accumulation of toxic metabolites. Nowadays, a crucial model of early diagnosed metabolic inherited disease, susceptible of a dietary and pharmacological specific therapy is phenylketonuria (PKU). PKU, identified in 1960 by Guthrie [1], was the first disease for which a simple test was developed to identify hyperphenylalaninemia (HPA) in large populations, thus becoming the first inborn metabolic disorder to benefit from newborn screening; its early detection and treatment demonstrated to prevent mental retardation [2-4].
Phenylketonuria (PKU) is a mendelian-inherited autosomal recessive inborn error of phenylalanine metabolism (prevalence of PKU 1:10,000) caused by deficiency or absence of the enzyme phenylalanine hydroxylase (PAH), that converts phenylalanine to tyrosine. In alternative it can be caused by defects involving the cofactor tetrahydrobiopterin (BH4), with a key role in this enzymatic step [5]. PAH gene is located on Chromosome 12 (12q23.2) and more than 560 mutations potentially involving PAH enzyme activity have been described. According to classical PKU classification, residual PAH activity and Phe blood concentration, correspond to different clinical types of PKU: classic, moderate and mild, with different dietary tolerance to Phe, so that free diet is possible in some patients. Classic PKU patients have Phe blood values > 1200 μM, while in moderate type Phe blood values range between 600 and 1200 μM, and in mild type between 360 and 600 μM [6]. New PKU guidelines classifies HPA as hyperpheylalaninemia characterized by Phe blood values between 120 and 360 μM (2-6 mg/dl) and Phe/ Tyrosine (Phe/Tyr) < 2,6 μM, and PKU, with Phe blood values > 360 μM and Phe/Tyr > 2,6 μM)[7,8]. If left untreated or late diagnosed, PKU results in increased phenylalanine concentrations in blood and brain (easily blood-brain-barrier), which cause severe irreversible intellectual disability, microcephaly, motor deficits, eczematous rash, autism, seizures, developmental problems, aberrant behavior and psychiatric symptoms.
We designed this study, with the idea that Phe/Tyr ratio could represent a more affordable marker of metabolic status in each patient, as well as his dietary and pharmacological treatments adherence than classic evaluation of Phe value only [9]. Moreover, a good Phe/Tyr ratio should mirror a good tyrosine amount both from conversion of phenylalanine and from free diet in patients with mild PKU, under Kuvan® treatment.
Patients and Methods
We present a retrospective, not-randomized, study of 15 patients, consecutively recruited into follow-up controls at our Center. They were 9 males (60%) and 6 females (40%), with age ranging from 4 to 39 years old (mean age 12.8 ± 9,92); 9 of them were younger than 12 years, and 6 older than 12 years. 13 patients are affected by classic PKU and undergo diet therapy (Amino acid supplementation/ Phe diet restriction) and 2 patients with mild PKU on a free diet and treated with sapropterin dihydrochloride (Kuvan®). Our patients series is composed by four pairs of brothers (8 patients) affected by classic PKU, on diet therapy, 5 patients with classic PKU on diet therapy (3 younger than 12 years and 2 > 12 years) and finally 2 patients (1 patient > 12 years and 1 patient < 12 years) with mild PKU, treated with Kuvan® and on a free diet. All patients were early diagnosed through newborn screenings and therefore all of them underwent early diet therapy, thus no one developed neuro cognitive delay
According to guidelines, Phe plasma levels under 12 years of age have to be maintained between 120 and 360 μM, while in patients aged >12 years Phe values should be between 360 and 600 μM [7,8]. All data came from DBS (dried blood spot) collected between December 2018 and April 2020 at follow-up controls of each patient.Phe, Tyr and ratio Phe/Tyr levels in DBS samples were measured using flow-injection electrospray–tandem mass spectrometry using QSight MD Mass Spectrometer with ESI source working in positive ion mode in Multiple reaction monitoring (MRM). In this way, through Non-derivatized MSMS Kit NeoBase2 (PerkinElmer QSight TM 210 MD), we carried out aminoacids dosage.
Result & Discussion
This preliminar data suggest that, into the same PKU type/ clinical phenotype and therefore Phe tollerance, exist different individual therapeutic responses. In all patients not fully adherent to diet therapy and/or pharmacological therapy, is very important to evaluate Phe/Tyr ratio, which value could be predictive of good or poor metabolic control, as well as being related to neurophysiological function [9]. Overall, in 10 patient of our series, a good correspondence was found between Phe and Phe/Tyr blood levels trend.
In Kuvan® responsive patients minor fluctuations were observed in blood Phe levels, in association with reduced Phe/Tyr ratio, predictive of neuroprotective action [10]. In particular, data analysis of first patient with mild PKU Kuvan® responsive (Table 1) show a perfect pharmacological treatment adherence, in fact the plasmatic values of Phe are always within the range of acceptability, compared to age (< 360 μM) and a good metabolic control or rather the ability to convert Phe in Tyr. The second mild PKU patient (Table 2) despite having a good adherence to drug treatment, as shown by plasmatic values of Phenylalanine within the therapeutic range (< 600 μM), present a poor metabolic control or rather a poor ability to convert phenylalanine into tyrosine as can be seen from the trend of the Phe/Tyr ratio, in the third and fifth sampling point, where the values of Phe/Tyr increase, compared to well controlled plasmatic phenylalanine value.
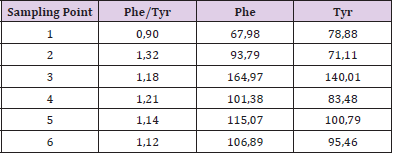
Table 1: First patient, 6 years old affected PKU Mild Kuvan® responsive and free diet.

Table 2: Second patient, male, 22 years old, affected PKU Mild Kuvan® responsive and free diet.
Regarding patients with classic PKU on a restrictive diet, all of them had plasmatic Phe values within the acceptable ranges compared to the age. From the evaluation of Phe/Tyr ratio trend it is clear that for plasmatic values of phenylalanine > 200 - 300 μM Phe/Tyr values increase significantly, indicating a reduced metabolic control or rather a reduced ability to convert Phe in tyrosine allowing to hypothesize the value of the Phe/Tyr ratio as a good marker of metabolic control. What has been said can be observed in the tables below relating two couples of siblings. The first pair of brothers (Tables 3 & 4) shows a very good adherence to diet therapy, as can be seen from plasmatic Phe concentration always within the acceptability range and a good metabolic control, intended as the ability to convert Phe to Tyr as demostred by Phe/ Tyr ratio trend.

Table 3: Brother and sister respectively 5 and 7 years old.
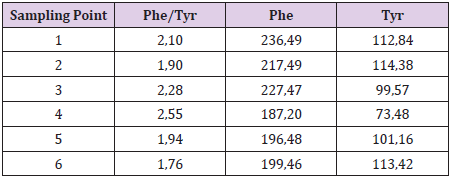
Table 4: Brother and sister respectively 5 and 7 years old.
Really interesting is the second couple (Tables 5 & 6). From the analysis of data, although they show overall good adherence to diet therapy, in fact plasmatic Phe values are below the therapeutic range (< 360 μM), from Phe/Tyr trend it is clear that for plasmatic values of phenylalanine greater than about 200 μM they can’t properly dispose of accumulated Phe converting it to Tyr, therefore their metabolic control worsens in fact Phe/Tyr ratio increases.
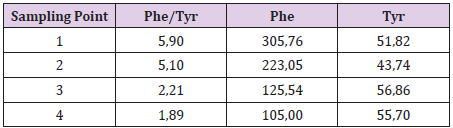
Table 5: Six-year-old twin brother and sister.
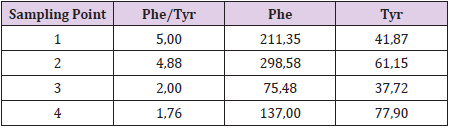
Table 6: Six-year-old twin brother and sister.
Conclusion
In our opinion, these preliminary data may suggest a possible prognostic role of Phe/Tyr ratio, for the evaluation of metabolic control and also diet and/or pharmacological treatment adherence more than classic evaluation of Phe value only. In particular, we think that Phe/Tyr ratio could give a more real idea of metabolic status in each patient. In fact, a good Phe/Tyr ratio should be the expression of a better diet therapy adherence in classic PKU patients, and of a good tyrosine amount (deriving both from conversion of Phe and from free diet in patients with mild PKU, on Kuvan® therapy). Our data suggest that, even into the same PKU type/clinical phenotype and therefore Phe tollerance, different individual therapeutic responses exist. We plan to continue this evaluation and this study, in a prospective analysis, to reach a very long time follow-up and a population study more representative. Another crucial role could be a closer control on diet and pharmacological therapy adherence by patients by an intensive and chronologically regular check of each patient. It is hoped to carry out an intra-family molecular study in particular for the couples of siblings involved in the study. We also plan, for all responsive Kuvan® patients, to carry out, on a scheduled follow up time, together with the dosage of Phe/Tyr ratio, also the dosage of the drug on DBS by tandem mass spectrometry, to compare the plasmatic concentration of the drug at each point of sampling.
Conflict of Interests
The authors declare no conflict of interest exist.
References
- Donald H Chace, William HHannon (2016) Technological Journey fromColorimetricto Tandem Mass Spectrometric Measurements in the Diagnostic Investigation for PhenylketonuriaJournal of Inborn Errors of Metabolism & Screening 4: 1-11.
- Blau N, Van Spronsen FJ, Levy HL (2010) Phenylketonuria. Lancet376(9750): 1417-1427.
- Hagedorn TS, Van Berkel P, Hammerschmidt G, Lhotáková M, Saludes RP, et al. (2013) Requirements for a minimum standard of care for phenylketonuria: the patient’s perspective. Orphanet J Rare Dis17(8):191.
- Hofman DL , Champ CL, Lawton CL, Henderson M, et al. (2018) A systematic review of cognitive functioning in early treated adults with phenylketonuria. Orphanet Journal of Rare Diseases 13: 150.
- Blau N, Hennemann JB, Langenbeck U, Lichter-Konecki U (2011) Diagnosis, classification, and genetics of phenylketonuria and tetrahydrobiopterin (BH4) deficiencies. Mol Genet Metab:104(Suppl): S2-9.
- Vockey J, Anderson HC, Antshel KM, Braveman NE, Burton BK, et al. (2014) Phenylalanine hydroxylase deficiency: diagnosis and management guideline. Genet Med 16(2): 188-200.
- Van Wegberg AMJ, MacDonald A, Ahring K, Bélanger-Quintana A, Blau N, etal. (2017) The complete European guidelines on phenylketonuria: diagnosis and treatment. Orphanet Journal of Rare Diseases12:162.
- Francjan J Van Spronsen, Annemiek MJ Van Wegberg, Kirsten Ahring, Amaya Bélanger-Quintana, Nenad Blau, et al (2017) Key European guidelines for the diagnosis and management of patients with phenylketonuria. Lancets Diabetes Endocrinol.
- Luciana M,Sullivan J, Charles ANelson (2001) Associations between phenylalanine to tyrosine ratios and performance on tests of neuropsychological function in adolescents treated early and continuously for phenylketonuria, Child. Dev. 72: 1637-1652.
- Anastasoaie V, Kurzius L, Peter Forbes, Susan Waisbren(2008) Stability of blood phenylalanine levels and IQ in children with phenylketonuria. MolGenetMetab 95: 17-20.
