 Review Article
Review ArticleAbstract
In March 2001, World Health Organization (WHO) Prequalification of Medicines Program (PQP) comes in action. A project partnered by United Nations (UN) and funded by The World Bank. This program prequalifies Active Pharmaceutical Ingredients (API), Finished Pharmaceuticals Products (FPPs) and Quality Control Laboratories (QCLs). API and FPP of certain therapeutic area that included in the WHO published Expression of Interest (EOI) for product evaluation list are under the scope of this PQP. All of these prequalification has to go through a series of steps which includes submission of a brunch of documents like- EOI, product dossier, site master file (SMF) etc., inspection of manufacturing site and clinical study center on case by case basis. The final outcome of this PQP is API and FPP listed in the WHO Public Assessment Reports (WHOPARs), manufacturing site and Contact Research Organizations (CROs) listed in the WHO Public Inspection Reports (WHOPIRs) and QCLs listed in the WHO prequalified quality control laboratories list.
Keywords: World Health Organization (WHO); Prequalification of Medicines Program (PQP); United Nations (UN); Active Pharmaceutical Ingredients (API); Finished Pharmaceuticals Products (FPPs); Quality Control Laboratories (QCLs); Expression of Interest (EOI); Site Master File (SMF); Contact Research Organizations (CROs); WHO Public Assessment Reports (WHOPARs); WHO Public Inspection Reports (WHOPIRs); Essential Medicines List (EML)
Introduction
Quality of medicines is always a problem in countries where regulatory and legal oversights are weak, medicines are not affordable to most and where often patients do not get medicines. To ensure effective treatment, to maintain patient and health-care worker confidence in treatment, quality is important. Additionally, it is also important to prevent development of resistance. As a realization of the fact, in 1977, the World Health Organization (WHO) published the first Model List of Essential Medicines (Essential Medicines List, EML). The EML assisted health authorities in selecting products for primary healthcare. It introduced the idea that some medicines are more important than others. Many later considered the first EML ‘a revolution in public health’ [1]. A project was introduced by United Nations (UN), managed by WHO to make an equally important decision to prequalify medicines in March 2001. World Bank funded this project. The primary objective of WHO Prequalification of Medicines Programme (PQP) is to contribute to the priority goal of United Nations which is to address common diseases in countries that have limited access to quality medicines [2]. Continuous improved quality of life saving medicines used by thousands of people in developing countries is the greatest accomplishment of this program.
Activities of Prequalification of Medicines Program (PQP)
There are three activities under this Prequalification of Medicines Program. These are:
a) Finished Pharmaceuticals Products (FPPs)/ Medicine Prequalification (2001)
b) Active Pharmaceutical Ingredients (API) Prequalification (2011)
c) Quality Control Laboratories (QCL) Prequalification (2004)
Prequalification of Medicines and Active Pharmaceutical Ingredients
First step of the prequalification process is the addition of a finished pharmaceutical product (FPP) or active pharmaceutical ingredient (API) in the invitation of manufacturers to submit an Expression of Interest (EOI) for Product Evaluation list. After discussion with WHO disease programmes and/or clinical specialists, this EOI is issued by WHO [3]. Generally, medicines that are identified as vital by WHO clinical experts for effective and development of treatment programmes or to safeguarding reproductive health are invited for evaluation. WHO Model List of Essential Medicines or Medicines for Children, and/or WHO treatment list or other relevant guidelines mainly enclosed all the medicines listed in the EOI. Although, there are some exceptions, such as medicine may not include in the model list of essential medicines and/or treatment guidelines are yet to be considered a vital for meeting treatment necessities and invited for assessment [3].
FPPs Eligible for Assessment [3]
Medicines of certain therapeutic class are mainly open for evaluation. Medicines of following therapeutic areas are currently open for invitations to manufacturers to submit an expression of interest (EOI).
a) Product of Diarrhoeal disease class.
b) Product for the treatment of Hepatitis B and C.
c) Product for the treatment of HIV/AIDS.
d) Product for the treatment of Influenza.
e) Product for the treatment of Malaria.
f) Product for the treatment of maternal and child health.
g) Product for the treatment of neglected tropical diseases.
h) Product for the treatment of reproductive health.
i) Product for the treatment of tuberculosis (Table 1).
APIs Eligible for Assessment [3]
APIs used to formulate medicines that are incorporated in the EOI or model lists of medicine are all qualified for prequalification, APIs of following therapeutic areas are eligible for assessment:
a) Product of Diarrheal disease class.
b) Product for the treatment of Hepatitis B and C.
c) Product for the treatment of HIV/AIDS.
d) Product for the treatment of Influenza.
e) Product for the treatment of Malaria.
f) Product for the treatment of maternal and child health.
g) Product for the treatment of neglected tropical diseases.
h) Product for the treatment of reproductive health.
i) Product for the treatment of tuberculosis.
Procedures for Prequalification
The following is a simplified overview of the APIs and FPPs prequalification process. Firstly, the API or FPP must be included in the relevant invitation to manufacturers to submit an EOI for Evaluation. Secondly, in accordance with the prequalification procedure manufacturer submits an application for the product (either API or FPP). After that, a screening checklist is used to check the submitted application for prequalification by WHO. They may ask for additional information after screening. Key elements of an evaluation of generic FPP are the insight of screening checklist. If the submitted dossier satisfies the screening requirements, then a reference number is assigned and accepted for full evaluation. Then, abbreviated assessment procedure may be followed by the applicant for the API or FPP that already been registered by stringent regulatory authority. Finally, in accordance with WHO good practices, dossier assessment and manufacturing compliance of the API and FPP and clinical testing sites verification are performed [4] (Figure 1).
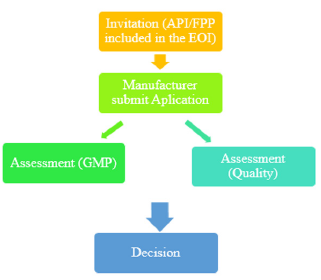
Figure 1: Prequalification Procedures.
Assessment Procedure of FPP
There are two procedures for FPP prequalification. Each procedure has specific submission requirements.
a) Full assessment and
b) Abbreviated assessment.
Full Assessment Procedures [5]
In order to be prequalified, a multisource (generic) finished pharmaceutical product (FPP) must meet the requirements relating to the Active Pharmaceutical Ingredient(s) (APIs) it contains, Multisource (generic) Finished Pharmaceutical Product (FPP) usually uses this process to prequalify. Generic products must meet all the requirements of active pharmaceutical ingredient(s) (APIs) use in the formulation, finished pharmaceutical product (FPP) requirement, and the GMP compliance requirement of site(s) at which the FPP is manufactured. This will include:
a) Bioequivalence study of the FPP.
b) Quality of the API(s).
c) Quality of the FPP.
d) WHO Good Manufacturing Practices compliance.
e) Inspection of that CRO with respect to the particular study performed and submitted in the dossier [6] (Figure 2).
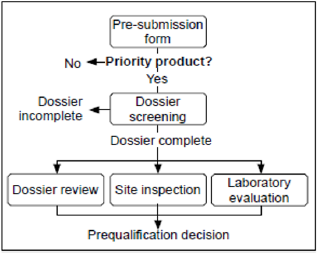
Figure 2: Full assessment [6].
Documents and Data to be Submitted [7]:
Usually, as specified in the invitations for EOIs, manufacturers of the FPPs are the applicants who participate in the WHO prequalification system for pharmaceutical products. In case where applicant is not the manufacturer of the product then contract manufacturing documentation, quality related documents and any other documents as ask by the reviewer need to be submitted. Product dossier with relevant information need to submit by the interested manufacturer to WHO principal point before the deadline mentioned in the invitation. WHO web site contained all the required submission guideline and instructions for dossiers. As a part of product evaluation, following documents need to be submitted to the WHO principal point:
a) Cover letter expressing interest of participation,
b) A product dossier,
c) Product samples,
d) site master file (SMF) of each manufacturing site, and
e) master file of clinical study site
English must be use for all submitted documentation.
a) Cover letter In the covering letter, manufacturers who are interested to prequalify their product, need to confirm that the information submitted in the product dossier is complete and correct.
b) Product Dossier [5]: Information attesting to the quality, safety and efficacy of the product must contain in the product dossier. Product dossier need to compile as per International Council on Harmonisation (ICH) modular format of the Common technical document (CTD) M4 Technical Requirements for Registration of Pharmaceuticals for Human Use. Dossier templates must be following for summarized information of:
a) Quality Information Summary (QIS)
b) Quality Overall Summary- Product dossier (QOS-PD)
c) BE trial information form (BTIF)
d) Biowaiver application form (If required)
API: Product dossier may contain any one of the following API information:
a) API listed in the WHO-prequalified list
b) A CEP issued by EDQM
c) Drug Master File (DMF) or API master file (APIMF)
or:
d) All completed section of module 3.2.S in the submitted FPP dossier for assessment.
“Guidelines on submission of documentation for a multisource (generic) finished pharmaceutical product for the WHO Prequalification of Medicines Programme: quality part, 2012” contain complete requirement of above discussed sections. Details procedure of API prequalification is provided later in this article.
Stability: Climatic Zone IV splitted into Zone IVa (hot and humid) with storage conditions of 30°C/65% RH and Zone IVb (hot and very humid) with storage conditions of 30°C/75% RH for the pharmaceutical preparation by WHO expert committee. Till September 2011, long term stability data at Zone IV conditions (30°C/65% for IVa or 30°C/75% for IVb, respectively) and in some cases, at Zone II (25°C/60%) data was accepted. Now-a-days, WHO expect all prequalified medicines will be used in all sub zones of III and IV unless justified by the applicant (Figure 3). Therefore, in order to maintain product quality throughout its entire shelf life, stability study need to be performed at climatic zones IVb. Based on complete long-term data at 30°C ±2°C/75% RH ±5% RH as required during dossier submission, the shelf-life should be established [8,9] (Figure 3).
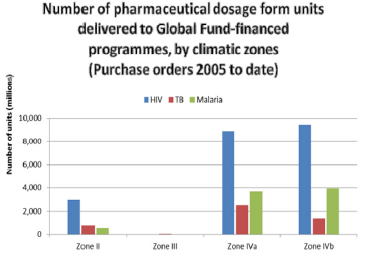
Figure 3: The number of units of HIV, TB and malaria products supplied to Zones II, III, IVa or IVb countries during the period 2007 – March 2010.
Bioequivalence [10]
Standards between Multisource (generic) products and originator or comparator products must be same. Generic manufacturer must ensure their products are of:
1. Same quality standards as those applicable to the comparator product
2. Therapeutically equivalent or bioequivalent to the comparator product
Following information of comparator product should include in the product dossier:
1. Origin (Country)
2. Expiry date and Lot/Batch number
3. Analysis results as a prove of pharmaceutical equivalence.
A list of comparator products recommended for use in bioequivalence study available in WHO web site. Stringent regulatory authority (SRA) market should be used to purchase comparator product. Currently, comparator products from ICH members such as European Union, Japan, Canada, USA, Switzerland and other ICH associated country like Norway, Australia, Liechtenstein and Iceland are accepted by WHO. In case, if no WHO-recommended comparator product can be identified or not available in the above mention markets, applicant should consult with WHO Prequalification Team: medicine regarding selection of comparator product. Any query related to bioequivalence study or comparator product should be sent to Dr Matthias Stahl at stahlm@ who.int. This question must be sent before initiation of study.
In addition to this, submitted product dossier must contain following comparator product documents
Labelling of comparator product
b) Invoice/ Proof of Purchase of comparator product with clearly visible distributor information
c) Method of shipment documentation
d) Storage conditions from the time of purchase to the time of study initiation
e) A statement-certifying the authenticity of the comparator product documents and that the comparator product was purchased from the specified national market.
Below BE study report should be incorporated in the product dossier:
a) Successful pivotal bioequivalence study report along with Bioequivalence Trial Information Form (BTIF) as per WHO guidance
b) Summary tables of all bioequivalence study conducted on same formulation and manufacturing process irrespective of study result in module 2.7 of the dossier
Agency may ask for the complete report if they think it is required to review.
c. Product Samples
Complete package of samples, for the assessment of appearance, material of container and product labelling. It permits reviewer to assess physical, chemical and pharmaceutical analysis [11].
d. Site Master File (SMF):
Site master file usually prepared by manufacturer with information of production and/or control of manufacturing operations at the said site and any closely operations performed at adjacent building. SMF must be compiled in English [11].
e. Contract Research Organization Master File (CROMF)
CROMF for each clinical study sites need to be submitted in the specified WHO format like SMF [11].
Abbreviated Assessment- SRA-Approved Multisource (Generic) or Innovator FPPs:
WHO accepts scientific assessment of medicinal products that has been carried out by stringent regulatory authorities that follow similar quality, safety and efficacy standard as recommended by WHO [12].
Stringent regulatory authority (SRA) is defined as a regulatory authority which is:
a) Member of the ICH; or
b) ICH observer, or
c) Authority allied with an ICH member through a legallybinding, mutual recognition agreement including Australia, Iceland, Liechtenstein and Norway (as may be updated from time to time) [13] (Figure 4).
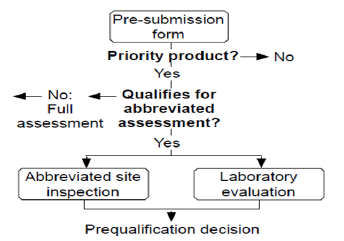
Figure 4: Abbreviated assessment [6].
Data and Information to be Submitted [13]
a) Cover letter
b) Marketing authorization, or the equivalent certificate issued by the SRA.
c) WHO-type certificate of a pharmaceutical product (COPP)
d) Current SRA-approved summary of product characteristics (SmPC), or an equivalent, patient information leaflet (PIL), or equivalent, and the labelling along with SRA-approved website link if available.
e) A list of the manufacturer(s) of the FPP approved by SRA with addresses,
f) A list of the manufacturer(s) of the API approved by SRA with addresses,
g) Any assessment report(s) issued by SRA if available.
h) Lot Distribution data in a tabular format that has been manufactured/commercialized in the reference SRA since approval or during last 05 years along with annual product quality review (APQR).
i) Product sample with certificate of Analysis (CoA).
j) Current release and shelf-life specifications of FPPs with method of analysis.
k) Fully completed quality information summary (QIS-SRA). QIS-SRA template, available at website (http://apps.who.int/ prequal/).
Submission Procedure [14]
It consists of two steps:
a) Step 1: Submission of documentation for the purpose of screening: Pictorial presentation of this step is provided in Figure 5.
b) Step 2: Document submission
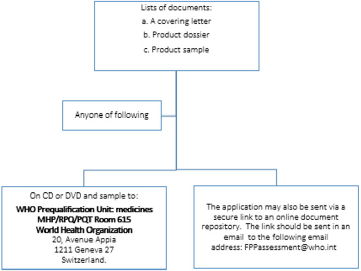
Figure 5: Step 1: Submission of documentation for the purpose of screening.
After applicant received a confirmation email or letter from WHO that, the dossier has been accepted for evaluation and a reference number assigned for its submission can be made [14]. Pictorial presentation of this step is provided in Figure 6.
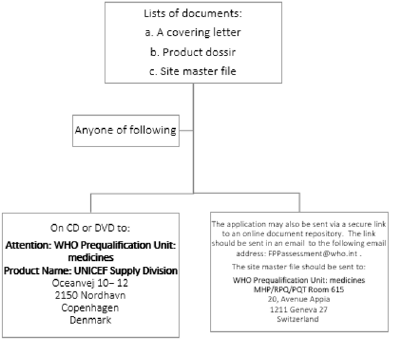
Figure 6: Step 2: Submission of documentation.
Assessment Procedure of API
APIs prequalification is an independent procedure that classifies APIs that are of good quality and manufactured in compliance with GMP standard. Prequalification of FPPs is greatly facilitated if a prequalified API is used to formulate the FPP [15].
There are two procedures
Full assessment for APIMF Not Previously Assessed
b) DMF/ API Master File (APIMF) Procedure.
Full Assessment for APIMF Not Previously Assessed [16]: If an API not previously assessed by SRA or WHO, it requires
a) Complete assessment of DMF/ APIMF
b) API manufacturing site inspection.
DMF/ API Master File (APIMF) Procedure [17]: Due to confidentiality, this procedure is increasingly popular now.
FPP applicant submit the open part (OP) of APIMF as part of the dossier along with a Letter of Access from API manufacturer that provide WHO prequalification team to review the restricted part (RP) of APIMF. FPP manufacturer need to ensure that the API manufacturer provide the required documents as requested by the agency for review. APIMF reassessment usually not required but “acceptability” of API manufacturer will always be reviewed as part of FPP prequalification (Figure 7).
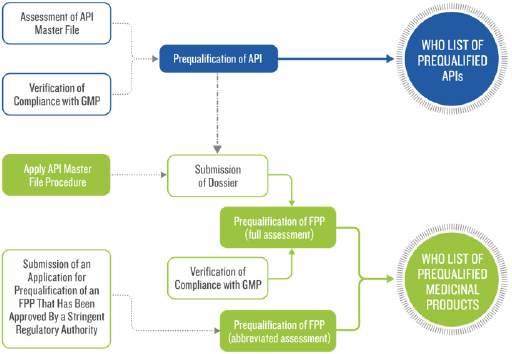
Figure 7: The number of units of HIV, TB and malaria products supplied to Zones II, III, IVa or IVb countries during the period 2007 – March 2010.
A. Prequalification of Quality Control Laboratories
To meet the standards of quality and safety of FPPs and APIs quality control laboratories plays a vital role. If a laboratory complies with WHO Good Practices for Quality Control Laboratories, it can get prequalified as QCL status [18].
Assessment Procedure of QCLs (Figure 8)
The process consists of the following steps:
a) Submit an Expression of Interest for Prequalification.
b) Submits an application -laboratory information file/ quality manual
c) Agency reviews laboratory information file/ quality manual may request additional information and schedule inspection of the QCL.

Figure 8: Procedure WHO prequalification Program for QCLs.
When a laboratory complies with all the prequalification requirements it will added in the WHO list of prequalified laboratories [4].
Below laboratory information file should be submitted and cover the areas:
a) General laboratory information with proposed activities for prequalification.
b) Inspections and external audits performed along with Quality management system.
c) Participation in proficiency tests and/or trials.
d) Internal audits.
e) Documentation record and controls.
f) Personnel.
g) Premises.
h) Equipment.
i) Lists of reference substances and reagents.
j) Testing performed as contract laboratory (if applicable).
k) Handling of samples.
l) Validation and/or verification of analytical procedures.
m) Investigation of out-of-specification (OOS) results.
n) Testing of stability sample (if applicable).
o) Testing of microbiology samples (if applicable) [19,20].
Site Inspection
Planning and coordination of site Inspection of API and FPP and clinical testing units or CROs will be done based on
a) SOPs.
b) Quality risk management (QRM) principles [7].
Outcome of the Prequalification Procedure
WHO will publish the evaluation report named “WHO Public Assessment Report (WHOPAR(s))” for the product dossier and inspection reports called “WHO Public Inspection Reports (WHOPIR(s))” for manufacturers of API & FPP and CROs, if they comply with the set standard [21].
Fees
From 1st January 2017, a new fee schedule has been published by WHO API, FPP and Vaccines. This new fee schedule will be monitored, and assessment will be done for 03-year time [22] (Table 2).
Prequalification Validity
Table 3 defines the validity period for prequalified Active Pharmaceutical Ingredients (API), Finished Pharmaceuticals Products (FPPs) and Quality Control Laboratories (QCLs) (Table 3).
Conclusion
Prequalification program of WHO is a renowned and broadly recognized tool, which acts as a self-regulating body and represents a sole global system. It ensures quality, safety and efficacy of priority medicines as per international and unified standards. The main role of WHO-PQP is to facilitate access for under privileged to get quality products. It also plays an active role to build up capacity with national regulators, QCLs and manufacturers.
Conflict of Interest
We have no disclosure to make that qualifies as a conflict of interest.
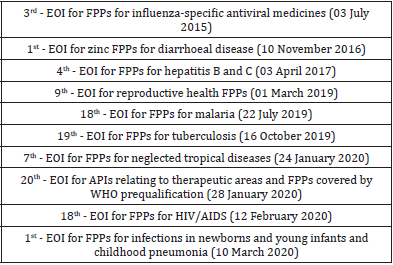
Table 1: Lists of published API and FPP invitations to manufacturers to submit an expression of interest (EOI).
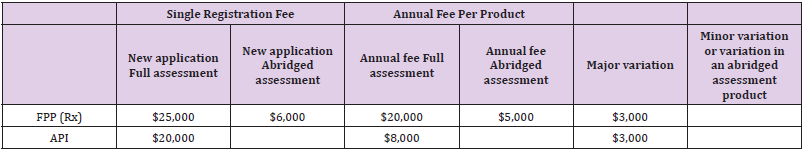
Table 2: levels of significant differences among proportions of surviving, average pupae production and fecundity G. pallidipes flies under feeding frequency tests analyzed using multi linear regression and Tukey mean.

Table 3: levels of significant differences among proportions of surviving, average pupae production and fecundity G. pallidipes flies under feeding frequency tests analyzed using multi linear regression and Tukey mean.
References
- Ellen FM Hoena, Hans V Hogerzeilb, Jonathan D Quickc, Hiiti B Sillod (2014) A quiet revolution in global public health: The World Health Organization’s Prequalification of Medicines Programme. Journal of Public Health Policy 35(2): 137-161.
- M Stahl (2013) The WHO Prequalification of Medicines Programme, CPTR 2013 Workshop, Washington, 1-3.
- FPPs & APIs Eligible for Prequalification ("EOIs").
- https://extranet.who.int/prequal/content/prequalification-procedures-and-fees-0
- https://extranet.who.int/prequal/content/full-assessment-multisource-generic-fpps-0
- (2014) WHO Drug Information. Bringing quality-assured in vitro diagnostics to WHO Member States. 28(3).
- (2011) WHO Technical Report Series. Annex 10: Procedure for prequalification of pharmaceutical products. WHO 96: 373-390.
- (2009) WHO Technical Report Series. Annex 2: Stability testing of active pharmaceutical ingredients and finished pharmaceutical products. WHO 953: 87-130.
- (2012) WHO Technical Report Series. Annex 4, 3.2.P.8: Guidelines on submission of documentation for a multisource (generic) finished pharmaceutical product for the WHO Prequalification of Medicines Programme: quality part. WHO 970.
- https://extranet.who.int/prequal/content/bioequivalence-0
- https://extranet.who.int/prequal/content/submission-procedure-expression-interest-eoi-full-assessment-multisource-generic-fpp
- https://extranet.who.int/prequal/content/abbreviated-assessment-multisource-generic-or-innovator-product-0
- (2014) WHO Technical Report Series. Annex 5 Guidelines on submission of documentation for prequalification of finished pharmaceutical products approved by stringent regulatory authorities. WHO 986: 313-316.
- https://extranet.who.int/prequal/content/submission-procedure-expression-interest-eoi-full-assessment-multisource-generic-fpp
- https://extranet.who.int/prequal/content/active-pharmaceutical-ingredients
- https://extranet.who.int/prequal/content/full-api-assessment-%E2%80%95-apimf-not-previously-assessed
- https://extranet.who.int/prequal/content/api-master-file-apimf-procedure
- https://extranet.who.int/prequal/information/quality-control-laboratories
- https://www.who.int/medicines/areas/quality_safety/quality_assurance/trs1003_annex3.pdf?ua=1
- (2011) WHO Technical Report Series. Annex 13: WHO guidelines for preparing a laboratory information file. World Health Organization 961: 403-408.
- (2004) World Health Assembly Resolution. Fifty-Seventh World Health Assembly.
- https://extranet.who.int/prequal/sites/default/files/documents/PQ_Fees_012018.pdf
