 Case Report
Case ReportAbstract
Background: The transcription factor ZFP42, located on chromosome 4q35, is a pluripotency marker in early embryonic stem cells. ZFP42 is expressed in embryonic stem cells and downregulated in early development after exposure to retinoic acid. We present a patient who developed a neuroblastoma and a secondary peripheral atypical cartilaginous tumor during childhood. She has a germline rearrangement involving a genomic region close to the ZFP42 gene.
Methods: Data was collected on clinical and tumor characteristics. (Shallow) whole genome sequencing (WGS) was performed on neuroblastoma and whole blood samples. ZFP42 expression of in the cartilage tumor was analyzed using RT-PCR.
Result: WGS revealed a germline rearrangement resulting in the duplication of a 403kbp region and the relocation of a 154kbp region, in between the duplicated regions, on chromosome 4q35. Analysis of publicly available ChIP-seq data of neuroblastoma cell lines revealed the presence of the enhancer marks H3K27ac and H3K4me1 in the 154kbp region that is relocated in the patient. Due to the relocation, this active region is now positioned closer to ZFP42, suggesting “enhancer hijacking”. In addition, ZFP42 was detected in the cartilage tumor. Since ZFP42 is involved in the repression of retinoic acid induced differentiation of embryonic stem cells, we hypothesize that the activation of ZFP42 in this patient resulted in a differentiation arrest and subsequent tumor susceptibility.
Conclusion: We present a patient, with a germline rearrangement upstream of ZFP42, who developed two neoplasms in childhood. We propose a role for ZFP42 as a tumor predisposition gene in this patient.
Keywords: Neuroblastoma; Chondrosarcoma; Germline; ZFP42; REX-1
Abbreviations: 123/131I-MIBG: Iodine- 123/131 Metaiodobenzylguanidine; CNV: Copy Number Variation; HVA: Homovanillic Acid; RT-PCR: Reverse Transcription Polymerase Chain Reaction; VMA: Vanillylmandelic Acid; WGS: Whole Genome Sequencing; ZFP42: Zinc Finger Protein 42
Introduction
Neuroblastoma is a developmental malignancy of the neural crest [1]. Tumors typically arise in young children, with a median age of 18 months at diagnosis, and most frequently reside in the adrenal gland or sympathetic side chain [2,3].The survival rates are high (>85% event free survival) for patients with localized disease, but low (<50%) for patients above 18 months with metastatic disease [2]. Most cases occur sporadically, but familial neuroblastoma is observed in 1-2% of patients [4]. Familial neuroblastoma cases are mostly (80%) attributed to ALK mutations [5,6]. The second most frequently mutated gene in familial neuroblastoma is PHOX2B, causing 6-10% of familial cases [7]. Genetic predisposition to neuroblastoma can also be caused by more general cancer predisposition syndromes, such as neurofibromatosis type 1 (NF1), Li-Fraumeni syndrome (TP53), Costello syndrome (HRAS), Noonan syndrome (PTPN11, KRAS), Beckwith-Wiedemann syndrome (chromosome 11) and Fanconi Anemia (FA pathway) [8].
Chondrosarcoma is a tumor showing cartilaginous differentiation. Chondrocytes, the cartilage producing cells, are partly neural crest derived. It is the second most frequent bone tumor after osteosarcoma, representing about 20% of bone tumors [9]. Tumors arise predominantly between the age of 20 and 60 years. Approximately 85% of chondrosarcomas are classified as conventional chondrosarcomas. These tumors develop in benign enchondromas (central chondrosarcoma) or osteochondromas (peripheral chondrosarcoma) [10]. However, only 1-3% of osteochondromas will develop into secondary peripheral chondrosarcomas [9]. Peripheral chondrosarcoma occurs in younger patients and generally behave less malignant than central chondrosarcomas [11,12]. Prognosis largely depends on histopathological grading based on tumor cell density, mitotic figures and cartilaginous matrix. Atypical cartilaginous tumors/ grade I chondrosarcomas, are regarded locally aggressive and rarely metastasize, the 5-yr disease specific survival is estimated at 100% and 10-yr overall survival at 83% [10,12,13]. Grade II and III tumors have higher cell densities and less matrix and 10-yr OS is 64% and 29%, respectively.10 Predisposition for the development of chondrosarcomas can be caused by germline inactivating mutations in the EXT1 or EXT2 genes, causing hereditary multiple osteochondromas [14-16]. Somatic mosaicism of mutations in IDH1 or IDH2 causes Ollier disease and Maffucci syndrome, which also predispose for development of chondrosarcoma [17,18]. In children, secondary peripheral chondrosarcomas are associated with radiation therapy [19].
ZFP42 is a zinc finger transcription factor, a YY1 transcription factor family member, located on chromosome 4q35, first recognized in 1989 [20,21]. It was identified by its downregulation shortly after treatment with retinoic acid (RA) in F9 teratocarcinoma stem cells, which resulted in differentiation of these cells [20]. ZFP42 was subsequently named REX-1 (for reduced expression). Although some aspects of the function of ZFP42 are still unclear, ZFP42 is used as a marker of pluripotency for embryonic stem cells and induced pluripotent stem cells. Expression in mouse and human is restricted to embryonic stem cells, tissue specific stem cells and germ cells of the testis [21-23]. A specific role for ZFP42 in cancer is unknown. Being a stem cell marker, it is believed to be expressed in cancer stem cells. Expression has been found in some carcinoma cell lines and in glioblastoma multiforme [20,21]. ZFP42 is positively regulated by Nanog, Sox2, Oct3/4 and Oct6, also known markers for pluripotent stem cells [24,25]. Here, we present a patient with two unrelated neoplasms in childhood, who carries a germline rearrangement, in a genomic region upstream of ZFP42.
Material and Methods
Patient
The patient’s chart was reviewed for: patient characteristics, symptoms at diagnosis, treatment, pathology reports, radiological and nuclear medicine reports, and urinary excretion levels of homovanillic acid (HVA) and vanillylmandelic acid (VMA).
Shallow and Whole Genome Sequencing
Shallow whole genome sequencing was performed on FFPE material of the neuroblastoma as described before [26]. DNA from a whole blood sample was isolated using the DSP DNA Midi kit according to manufacturer’s protocol (Qiagen, Venlo, The Netherlands). Library preparation and sequencing were performed on the HiSeqX sequencing system (Illumina, San Diego, CA, USA), the BCL output was converted using the bcl2fastq tool (Illumina, v.2.20, San Diego, CA, USA) as described before [27]. For analysis and visualization, the “R2: Genomics Analysis and Visualization Platform (http://r2.amc.nl)” was used.
Cartilage Tumor RNA Isolation
Twenty μm section of frozen tissue were collected in a tube and dissolved in 5 ml TRIzol. Homogenization was performed using the Ultra-Turrax (IKA, Staufen, Germany). After mixing for 1 min. with chloroform (1 ml), the sample was centrifuged for 30 min. at 15.000xg. A second phenol/chloroform extraction was performed similarly, followed by isopropanol precipitation. The RNA was dissolved in 30 μl MQ. cDNA was synthesized using RevertAid H Minus FirstStrand cDNA synthesis Kit (Thermo Fisher) according to the recommendations of the manufacturer (372 ng RNA per 20 μl reaction).
RT-PCR and Gel Electrophoresis
Reverse transcriptase PCR (RT-PCR) was performed on cDNA using ReddyMix PCR Mastermix (ThermoFisher), and forward and reverse primers at 500 nM. Forward and reverse primer sequence of ZFP42 were GGCCTTCACTCTAGTAGTGCTC and CTGGCTCATGTTTTCCTGCC, respectively. UBC was used as control gene to ensure cDNA quality. Forward and reverse primer sequence of UBC were ATTTGGGTCGCGGTTCTTG and TGCCTTGACATTCTCGATGGT, respectively. The reaction was performed with heating for 3 min at 95°C, followed by 40 cycles of 30 sec. at 95°C, 30 sec. at 60°C and 30 sec. at 70°C. Subsequently, RT-PCR products were visualized with gel electrophoresis.
Ethics
Written informed consent for the use of stored material was obtained from the patient.
Case Report
In 1997, a 15-months old girl was diagnosed with neuroblastoma stage 4 in the Democratic Republic of the Congo and the primary tumor in the left adrenal gland was resected. In 2000, the patient immigrated to the Netherlands, still suffering from Iodine-123 Metaiodobenzylguanidine (123I-MIBG) avid metastatic disease (Figures 1A & 1B), histology of a lymph node metastasis showed ganglioneuroblastoma morphology. After 1.5 years of wait-and-see, a 123I-MIBG-avid meningeal metastasis showed progression. Chemotherapy was started, followed by 131I-MIBG therapy, but all metastases were unresponsive. Next, treatment with isotretinoin (13-cis retinoic acid) was administered in an attempt to cause differentiation of the metastases. Chemotherapy resistance in patients with metastatic neuroblastoma is associated with a poor prognosis [28]. However, in this patient, no progression was observed, but a very slow metabolic response was observed by a decrease but not normalization in 123I-MIBG uptake (Figures 1C & 1D) and normalization of HVA 6.25 years after the initial diagnosis. This is an unusual course of disease for a neuroblastoma, as aggressive growth with fatal outcome was expected
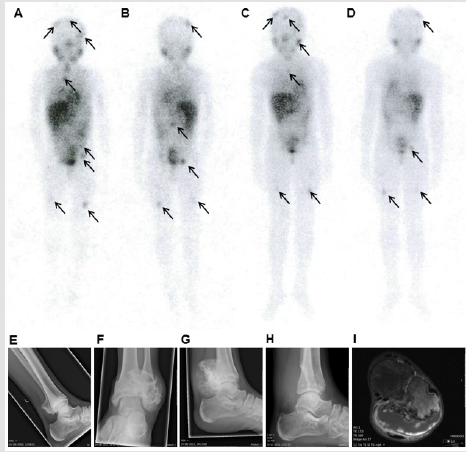
Figure 1: Radiological and nuclear imaging of the patient.
Note: Figure 1 (A-D) whole body 123I-MIBG single photon emission computed tomography (SPECT) scans, the arrows indicate pathological 123I-MIBG uptake. A/B) anterior and posterior scans 3 years after diagnosis; C/D) anterior and posterior scans, 6 years after diagnosis. Figure 1 (E-H): conventional X-rays of the left ankle showing: E) an osteochondroma of the distal fibula at the age of 12 years, F/G) anterior/posterior and lateral X-ray at the age of 16 years, showing fast progression of the osteochondroma . H) lateral X-ray of the left ankle after resection of the cartilage tumor. I) T1 weighted MRI at the age of 16 years, showing an osteochondroma of the distal fibula metaphysis with a cartilage cap of 2.6 cm maximum diameter and gadolinium enhancement at the border of the cartilage cap, suspected for secondary peripheral chondrosarcoma development.
At the age of 12 years, the patient developed an osteochondroma, a benign cartilage tumor, at the surface of the left distal fibula (Figure 1E). Over the period of four years this tumor developed into a secondary peripheral chondrosarcoma/atypical cartilaginous tumor as the size of the cartilaginous cap exceeded 2 cm (Figures 1F-1I) [9,29]. Development of a secondary peripheral atypical cartilaginous tumor is exceptional for this age, especially since the patient was not treated with radiotherapy for the neuroblastoma [16,30]. The cartilage tumor was surgically resected (R0 resection) (Figure 1H), no adjuvant chemotherapy was administered. At last follow-up, 20 years after the neuroblastoma diagnosis and 4 years after the secondary peripheral atypical cartilaginous tumor diagnosis, the patient was alive and well. Because the patient developed two different neoplasms during childhood and because her neuroblastoma followed an aberrant indolent clinical course, we tested for germline aberrations.
WGS of Germline DNA Identifies a Relocation and Duplication Near ZFP42
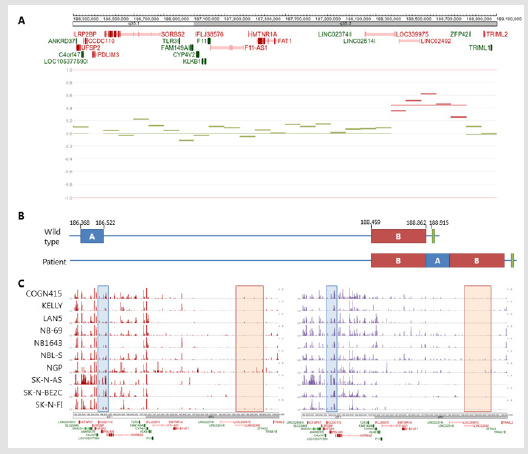
Figure 2: Germline rearrangement.
Note: Figure 2A) Copy number variation (CNV) plot of chr4q35 of the patient in the shallow WGS neuroblastoma sample, showing a duplication in front of ZFP42. 2B) schematic overview of ZFP42 in normal configuration (upper overview) and as found in the germline of the patient (lower overview), blue is the relocated region (A), red the duplicated region (B), green the ZFP42 gene. Chromosomal localization is indicated in kbp resolution. 2C) ChIP-seq results of H3K27ac (red peaks, left) and H3K4me1 (purple peaks, right) in neuroblastoma cell lines (GSE138314 dataset). The blue box indicates the relocated region, the red box the duplicated region.
Whole genome sequencing (WGS) on whole blood of the patient, revealed a germline heterozygous rearrangement on chromosome 4q (Figure 2). In the rearrangement, a 403 kbp region was duplicated and a 154 kbp region was relocated in between the duplicated regions (Figures 2A & 2B). The duplicated region contained no protein coding genes. The relocated region caused breakpoints in the C4orf47, CCDC110, and SORBS2 genes. These genes are not known as tumor suppressor genes. Interestingly, the rearrangement was located near the ZFP42 gene, a pluripotency marker for embryonic stem cells. Due to lack of parental blood samples, it could not be established if the rearrangement was inherited or de novo. No other germline alterations of relevance were identified. Shallow whole genome sequencing of the neuroblastoma tumor showed a flat copy number variation plot, with the germline duplication on chromosome 4q35 as only structural aberration (Figure 2A). This could either indicate that the tumor content was too low, or that no other chromosomal aberrations were present in the neuroblastoma.
Next, we investigated the epigenetic status of the rearranged regions in a publicly available ChIP-seq dataset (GSE138314) of neuroblastoma cell lines for the enhancer marks H3K27ac and H3K4me1 using the R2 genomics analysis and visualization platform (http://r2.amc.nl). ChIP-seq data of these neuroblastoma cell lines revealed that the 403 kbp region that is duplicated in this patient had near absent levels of H3K27ac and H3K4me1 histone modification marks. However, the 154 kbp region that is relocated in this patient showed higher levels of H3K27ac and H3K4me1 in these cell lines (Figures 2B & 2C). Due to the relocation, a region positive for enhancer marks was positioned near the promoter of ZFP42 in this patient. Therefore, we hypothesized that this could result in activation of ZFP42, which is normally not expressed, by enhancer hijacking.
ZFP42 is Expressed in Chondrosarcoma of the Patient
Neuroblastoma tissue was not available for expression analysis of ZFP42. Therefore, we tested ZFP42 expression in the cartilage tumor. From the fresh frozen tumor tissue RNA was isolated and ZFP42 expression was measured using reverse transcriptase polymerase chain reaction (RT-PCR). With RT-PCR, expression of ZFP42 was found in the cartilage tumor (Figure 3). However, expression was low compared to the D341med medulloblastoma cell line, which served as a positive control. Sanger sequencing of the RT-PCR product confirmed the ZFP42 sequence.
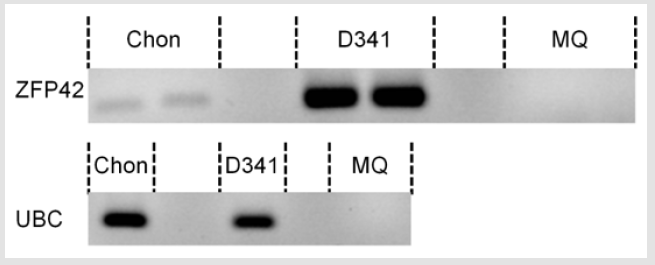
Figure 3: ZFP42 expression in the cartilage tumor.
Note: Gel electrophoresis of the RT-PCR product of the ZFP42 gene (top, in duplo) and of the UBC housekeeping gene (bottom), performed on the cartilage tumor (chon), D341med cell line (positive control) and MQ (negative control).
Discussion
We present a patient who developed two neoplasms in childhood, a disseminated neuroblastoma at 1 year of age with an unexpected indolent course of disease and a secondary peripheral atypical cartilaginous tumor at 16 years of age. We identified a germline rearrangement in chromosome 4q 35, resulting in the positioning of a region positive for enhancer marks near the promoter of the ZFP42 gene, a human stem cell marker [31,32]. ZFP42 was expressed in the cartilage tumor. The combination of two childhood neoplasms, the aberrant course of neuroblastoma disease, and the germline rearrangement affecting a pluripotency marker suggests that the ZFP42 gene is involved in the development of these tumors.
ZFP42 is a stem cell marker which is normally downregulated
in early development and which is overexpressed in glioblastoma
multiforme and invasive cervical squamous cell carcinoma [31-34].
In glioblastoma downregulation of ZFP42 resulted in a decreased
proliferation and increased differentiation.34 However, this result
was not confirmed in embryonic stem cells [32,35]. In embryonic
stem cells, ZFP42 had a role in inhibiting retinoic acid induced
differentiation. Knock-out of ZFP42 resulted in higher degree of
differentiation and growth inhibition after treatment with retinoic
acid. Overexpression of ZFP42 resulted in less differentiation and
an increase in growth [22]. Neuroblastomas are sensitive to retinoic
acid, which is used as differentiation treatment in patients [36-38].
Chondrosarcomas undergo growth inhibition after treatment with
retinoic acids or selective agonists of RARγ (retinoic acid receptor
gamma) in vitro and in vivo [39-42]. However, retinoic acid is not
part of the standard chondrosarcoma treatment protocol, as it is in
high-risk neuroblastoma treatment [37,39].
Neuroblastoma is a developmental tumor of the neural crest.
Tumors arise from a common precursor of the adrenal chromaffin
cells and sympathetic neurons. Chondrosarcomas develop from cells
committed to cartilaginous differentiation, which are partly neural
crest derived and partly mesoderm derived. The precursors of both
tumor types are influenced by retinoic acid for differentiation [43].
Based on literature, we propose that ZPF42 inhibits the effect of
retinoic acid on differentiation, resulting in a differentiation arrest
in the precursor cells. Avoiding differentiation is a crucial step in
cancer development [44,45]. However, more research is needed to confirm the role of ZFP42 in the inhibition of differentiation and the
potential role in cancer development.
Conclusion
In conclusion, the patient presented here developed two neoplasms in childhood, a neuroblastoma and a cartilage tumor, of which the precursor cells depend on retinoic acid for differentiation. The patient has a germline mutation affecting ZFP42, a repressor of retinoic acid induced differentiation. We propose a role for ZFP42 as a tumor predisposition gene in this patient.
Acknowledgement
This research was funded by the Villa Joep foundation.
Conflict of Interest
All authors declare to have no conflicts of interest
References
- Nakagawara A, Ohira M (2004) Comprehensive genomics linking between neural development and cancer: neuroblastoma as a model. Cancer Lett 204(2): 213-224.
- Cohn SL, Pearson AD, London WB, Tom Monclair, Peter F Ambros, et al. (2009) The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol 27(2): 289-297.
- Vo KT, Matthay KK, Neuhaus J, Wendy B London, Barbara Hero, et al. (2014) Clinical, biologic, and prognostic differences on the basis of primary tumor site in neuroblastoma: a report from the international neuroblastoma risk group project. J Clin Oncol 32(28): 3169-3176.
- Kushner BH, Gilbert F, Helson L (1986) Familial neuroblastoma. Case reports, literature review, and etiologic considerations. Cancer 57(9): 1887-1893.
- Janoueix-Lerosey I, Lequin D, Brugieres L, Agnès Ribeiro, Loïc de Pontual, et al. (2008) Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 455(7215): 967-970.
- Mosse YP, Laudenslager M, Longo L, Kristina A Cole, Andrew Wood, et al. (2008) Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 455(7215): 930-935.
- Trochet D, Bourdeaut F, Janoueix-Lerosey I, Anne Deville, Loïc de Pontual, et al.(2004) Germline mutations of the paired-like homeobox 2B (PHOX2B) gene in neuroblastoma. Am J Hum Genet 74(4): 761-764.
- Barr EK, Applebaum MA (2018) Genetic Predisposition to Neuroblastoma. Children (Basel) 5(9).
- Fletcher CD, Bridge JA, (editors) HPea. (2013) Chondrosarcoma (grades I-III), including primary and secondary variants and periosteal chondrosarcoma. In: World Health Organization classification of tumours. Pathology and genetics of tumours of soft tissue and bone, (4thEdn,), IARC Press, Lyon.
- Gelderblom H, Hogendoorn PC, Dijkstra SD, Carla S van Rijswijk, Augustinus D Krol, et al. (2008) The clinical approach towards chondrosarcoma. Oncologist 13(3): 320-329.
- Nota SP, Braun Y, Schwab JH, Van Dijk CN, Bramer JA (2015) The Identification of Prognostic Factors and Survival Statistics of Conventional Central Chondrosarcoma. Sarcoma 623746.
- Donati D, El Ghoneimy A, Bertoni F, Di Bella C, Mercuri M (2005) Surgical treatment and outcome of conventional pelvic chondrosarcoma. J Bone Joint Surg Br 87(11): 1527-1530.
- Van PraagVeroniek VM, Rueten-Budde AJ, Ho V, PDS Dijkstra, MAJ Van De Sande, et al. (2018) Incidence, outcomes and prognostic factors during 25 years of treatment of chondrosarcomas. Surg Oncol27(3):402-408.
- Heddar A, Fermey P, Coutant S, Emilie Angot, Jean-Christophe Sabourin, et al. (2017) Familial solitary chondrosarcoma resulting from germline EXT2 mutation. Genes Chromosomes Cancer56(2):128-134.
- Bovee JV (2008) Multiple osteochondromas. Orphanet J Rare Dis3:3.
- De Andrea CE, Reijnders CM, Kroon HM, D De Jong, P C W Hogendoorn, et al. (2012) Secondary peripheral chondrosarcoma evolving from osteochondroma as a result of outgrowth of cells with functional EXT. Oncogene31(9):1095-1104.
- Amary MF, Damato S, Halai D,Fitim Berisha, Fiona Bonar, et al.(2011) Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2. Nat Genet43(12):1262-1265.
- Pansuriya TC, Van Eijk R, D'Adamo P, Maayke A J H Van Ruler, Marieke L Kuijjer, et al. (2011) Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome. Nat Genet43(12):1256-1261.
- Huvos AG, Woodard HQ, Cahan WG, N L Higinbotham, F W Stewart, et al. (1985) Postradiation osteogenic sarcoma of bone and soft tissues. A clinicopathologic study of 66 patients. Cancer 55(6):1244-1255.
- Hosler BA, LaRosa GJ, Grippo JF, Gudas LJ (1989) Expression of REX-1, a gene containing zinc finger motifs, is rapidly reduced by retinoic acid in F9 teratocarcinoma cells. Mol Cell Biol9(12):5623-5629.
- Mongan NP, Martin KM, Gudas LJ (2006) The putative human stem cell marker, Rex-1 (Zfp42): structural classification and expression in normal human epithelial and carcinoma cell cultures. Mol Carcinog45(12):887-900.
- Scotland KB, Chen S, Sylvester R, Gudas LJ (2009) Analysis of Rex1 (zfp42) function in embryonic stem cell differentiation. Dev Dyn238(8):1863-1877.
- Bao L, He L, Chen J, Zhao Wu, Jing Liao, et al. (2011) Reprogramming of ovine adult fibroblasts to pluripotency via drug-inducible expression of defined factors. Cell Res21(4):600-608.
- Hamazaki T, Kehoe SM, Nakano T, Terada N (2006) The Grb2/Mek pathway represses Nanog in murine embryonic stem cells. Mol Cell Biol26(20):7539-7549.
- Shi W, Wang H, Pan G, Geng Y, Guo Y, et al. (2006) Regulation of the pluripotency marker Rex-1 by Nanog and Sox2. J Biol Chem281(33):23319-23325.
- Smeets SJ, Harjes U, Van Wieringen WN, Wessel N Van Wieringen, Daoud Sie, et al. (2011) To DNA or not to DNA? That is the question, when it comes to molecular subtyping for the clinic! Clin Cancer Res17(15):4959-4964.
- Priestley P, Baber J, Lolkema MP, NeeltjeSteeghs , Ewart De Bruijn, et al. (2019) Pan-cancer whole-genome analyses of metastatic solid tumours. Nature575(7781):210-216.
- London WB, Bagatell R, Weigel BJ, Elizabeth Fox,Dongjing Guo, et al. (2017) Historical time to disease progression and progression-free survival in patients with recurrent/refractory neuroblastoma treated in the modern era on Children's Oncology Group early-phase trials. Cancer123(24):4914-4923.
- De Andrea CE, Kroon HM, Wolterbeek R, Salvatore Romeo, Andrew E Rosenberg, et al. (2012) Interobserver reliability in the histopathological diagnosis of cartilaginous tumors in patients with multiple osteochondromas. Mod Pathol25(9):1275-1283.
- Wu AM, Li G, Zheng JW, D Chen, Z-G Qiao, et al. (2019) Chondrosarcoma in a paediatric population: a study of 247 cases. J Child Orthop13(1):89-99.
- Boroviak T, Loos R, Bertone P, Smith A, Nichols J (2014) The ability of inner-cell-mass cells to self-renew as embryonic stem cells is acquired following epiblast specification. Nat Cell Biol16(6):516-528.
- Meek S, Wei J, Oh T, Tom Watson, Jaime Olavarrieta, et al. (2020) A Stem Cell Reporter for Investigating Pluripotency and Self-Renewal in the Rat. Stem Cell Reports14(1):154-166.
- Zeng YT, Liu XF, Yang WT, Zheng PS (2019) REX1 promotes EMT-induced cell metastasis by activating the JAK2/STAT3-signaling pathway by targeting SOCS1 in cervical cancer. Oncogene38(43):6940-6957.
- Kim BS, Kang KS, Choi JI, Jung JS, Im YB, et al. (2011) Knockdown of the potential cancer stem-like cell marker Rex-1 improves chemotherapeutic effects in gliomas. Hum Gene Ther22(12):1551-1562.
- Masui S, Ohtsuka S, Yagi R, Takahashi K, Ko MS, et al. (2008) Rex1/Zfp42 is dispensable for pluripotency in mouse ES cells. BMC Dev Biol8:45.
- Peinemann F, Van Dalen EC, Tushabe DA, Berthold F (2015) Retinoic acid post consolidation therapy for high-risk neuroblastoma patients treated with autologous hematopoietic stem cell transplantation. Cochrane Database Syst Rev1:CD010685.
- Matthay KK, Villablanca JG, Seeger RC, D O Stram, R E Harris, et al. (1999) Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children's Cancer Group. N Engl J Med341(16):1165-1173.
- Sidell N (1982) Retinoic acid-induced growth inhibition and morphologic differentiation of human neuroblastoma cells in vitro. J Natl Cancer Inst68(4):589-596.
- Shield WP, Cellini A, Tian H, Dan Y, Joshua M Abzug, et al. (2020) Selective Agonists of Nuclear Retinoic Acid Receptor Gamma Inhibit Growth of HCS-2/8 Chondrosarcoma Cells. J Orthop Res38(5):1045-1051.
- Ettlin R, Galli B, Kistler A (1982) Histological changes during regression induced by retinoic acid in a transplantable rat chondrosarcoma. Virchows Arch APatholAnatHistol396(1):1-8.
- Oegema TR, Parzych SM (1981) Effect of the retinoic acid analog Ro 11-1430 on proteoglycans of swarm rat chondrosarcoma. J Natl Cancer Inst67(1):99-106.
- Thein R, Lotan R (1982) Sensitivity of cultured human osteosarcoma and chondrosarcoma cells to retinoic acid. Cancer Res42(11):4771-4775.
- Li X, Schwarz EM, Zuscik MJ, Randy N Rosier, Andreia M Ionescu, et al. (2003) Retinoic acid stimulates chondrocyte differentiation and enhances bone morphogenetic protein effects through induction of Smad1 and Smad5. Endocrinology. 144(6):2514-2523.
- Pierce GB, Speers WC (1988) Tumors as caricatures of the process of tissue renewal: prospects for therapy by directing differentiation. Cancer Res48(8):1996-2004.
- Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell100(1):57-70.
