 Review Article
Review ArticleAbstract
Protein phosphorylation is a reversible post-translational modification that involves a series of sequence-specific kinases and occurs on specific residues such as serine, threonine, and tyrosine. The reversible phosphorylation of proteins regulates almost all aspects of the cell’s life cycle and abnormal phosphorylation is the cause or consequence of many diseases. Protein phosphorylation states can mediate protein complex formation and regulate protein function, which is important for cell physiology but can also promote neuropathic events. The tau protein is a very important microtubule-associated protein in the brain, occurring most commonly in neurons and glial cells. Its level of phosphorylation is associated with a variety of diseases of the central nervous system such as Alzheimer’s disease. Under normal circumstances, post-transcriptional tau phosphorylation is conducive to the stability of microtubules. However, hyperphosphorylation can lead to the deformation and aggregation of various types of cytoskeletal components of nerve tissue, causing them to lose normal function.
Keywords: Protein; Tau Phosphorylation; Alzheimer’s Disease; Tubulin Structure; Drug Effects
Abbreviations: MAPS: Microtubule-Associated Proteins; AD: Alzheimer’s Disease; MTRS: Mercuric Transport; SER: Serine; THR: Threonine; MT: Microtubule; GSK3β: Glycogen Synthase Kinase 3β; ALA: Alanine; PD: Projection Domain; MBD: Microtubule- Binding Domain; PSP: Progressive Supranuclear Palsy; MAP1B: Microtubule Associated Protein 1B; GSK3: Glycogen Synthase Kinase 3; CDK5: Cyclin-Dependent Kinases 5; NFTs: Neurofibrillary Tangles; PHFs: Paired Helical Filaments; SFs: Straight Filaments; FTLD: Frontotemporal lobar degeneration; CJD: Creutzfeldt-Jakob Disease; PDPK: Proline- Dependent Protein Kinase; UPS: Ubiquitin-Proteasome System; PHF: Pair of Helical Filaments; PTPs: Protein Tyrosine Phosphatases; PSPs: Protein Serine/Threonine Phosphatases; PP1: Phosphatase Types 1; PsP2A: phosphatase types2A; PP2B: Phosphatase Types2B; PP2C: Phosphatase Types2C; PP: Protein Phosphatase; CCH: Chronic Cerebral Hypoperfusion; BRET: Bioluminescent Resonance Energy Transfer; PKA: Protein Kinases A
Introduction
Cellular proteins that bind to microtubules are collectively referred to as microtubule-associated proteins (MAPs). Under normal circumstances, MAPs are essential components for Maintaining the Structure and function of microtubules. They can increase the stability of microtubules, promote microtubule assembly, and regulate the relationship between microtubules and other cellular components [1]. MAPs con tain two functional regions: an alkaline binding domain that binds to the side of the microtubule; and an acidic salient binding domain that is an outwardly protruding filamentous structure in the form of a horizontal bridge connecting the MAP to other cell components, cytoskeleton components, membranes, and other structures. MAPs have microtubule binding activity, and their function can be performed by regulating the phosphorylation and dephosphorylation of specific amino acids.A variety of functions of MAPs involving the regulation of microtubule cytoskeletal dynamics have been discovered. MAPs are present in nerve tissue during neuronal development and play an indispensable role in microtubule remodeling during neuronal activity and in the stability of microtubules during neuronal maintenance. As a result, mutations in MAPs lead to neurodevelopmental disorders, psychiatric disorders, and neurodegenerative diseases.
MAPs are post-translationally regulated by phosphorylation, which can affect microtubule affinity, cell localization, or the overall function of specific MAPs with a profound effect on neuronal health. The microtubule-binding activity of a MAP is regulated by the phosphorylation and dephosphorylation of specific amino acids. The MAP family mainly includes MAP1, MAP2, tau, and MAP4. The first three are found mainly in neurons, while MAP4 exists in all kinds of cells. Of these four MAPs, the role of tau protein phosphorylation and dephosphorylation in Alzheimer’s Disease (AD) is the most extensively studied and significant progress has been made. The function of the microtubule-associated protein tau is to promote microtubule assembly and stabilization in neurons, which is required for axonal transport and neurite outgrowth [2]. Tau is a microtubule-associated phosphoprotein that is abundant in neurons and is regulated by protein kinases and protein phosphatases. Appropriately phosphorylated Tau binds to microtubules, thereby stimulating the assembly of tubulin into microtubules and maintaining microtubule stability [3]. In the brain of Alzheimer’s disease, tau is abnormally hyperphosphorylated; it contains three to four times more phosphate than normal tau [4]. In vitro and in vivo, hyperphosphorylation of tau has been shown to reduce the affinity of tau for microtubules, leading to disruption of neuronal cytoskeleton and axonal transport [5]. Abnormal aggregation of hyperphosphorylated tau protein is a common pathological feature of neurodevelopmental disorders commonly referred to as tauopathy, including AD, progressive supranuclear palsy and frontotemporal dementia [6]. Several neurodegenerative diseases, collectively referred to as tauopathy, are characterized by insoluble, highly phosphorylated tau that is a neuronal inclusion of straight or paired helical filaments [7].
Microtubules and the Effects of Tau Phosphorylation on Microtubule Structure
Neuronal development and function are influenced by the cytoskeletal infrastructure of cells, namely microtubules, actin, and intermediate filament networks. Microtubule cytoskeletal networks are organized into stable and dynamic arrays that provide structural support as molecular motion trajectories and serve as signal platforms during neuronal development and plasticity [8-10]. Microtubules are composed of alpha- and beta-tubulin heterodimers that assemble into protofilaments and then laterally contact each other to form tubules [11]. β-Tubulin must be in a GTP-bound state to allow the assembly of heterodimers onto the protofilament. Alpha- tubulin binds to β-tubulin but only β-tubulin can hydrolyze GTP. Once the protofilament is assembled, β-tubulin is exposed at the “plus end” and alpha-tubulin is exposed at the “minus end.” This structural polarity leads to a difference in the growth rate at each end and it has been observed that end-capping occurs more often [12] and is much faster on the plus end than on the minus end. Microtubules can be modified within cells by switching between assembled and disassembled states in a process called dynamic instability [13]. MAPs have the ability to bind to microtubule lattices, tubulin heterodimers, or both. They can thereby regulate the assembly/ disassembly kinetics of microtubules to properly organize and remodel microtubule cytoskeletal structure during neuronal development and activity [14,15].
The α- and β-tubulin heterodimers that assemble into microtubules exist in a state of dynamic equilibrium with non-polymeric tubulin. The filamentous structure of microtubules forms intracellular cytoskeletons in a variety of cells but are particularly enriched in neurons [16-18]. The dynamics of microtubule assembly can be regulated by temperature, microtubule protein modifications, small molecules such as paclitaxel, and some mercuric transport (MerT) interacting proteins [19-21]. Since microtubules play an important role in a wide range of biological functions, including the structural formation of neurons and the transport of intracellular substances, it is speculated that microtubule disruption (if any) can profoundly influence neuronal structure and function [22-24]. Tau protein has been identified as a factor that promotes microtubule assembly and stability. Microtubule assembly is thought to be negatively regulated by tau protein phosphorylation. More than 40 serine (Ser) and Threonine (Thr) residues have been identified as possible phosphorylation sites on the tau protein. Although the biological significance of every single phosphorylation site is not clear, it is known that phosphorylation of tau at Ser-262 of tau (in the 441-residue tau protein) has a profound influence on its interaction with microtubules [25].
Effect of Tau Phosphorylation on the Structure of Threonine 231
The amino acid sequence that interacts with MT in Tau is localized to a proline-rich region and a repeat domain. Tau contains 85 potential phosphorylation sites, of which three sites S214, T231 and S262 are critical for Tau-MT interaction. In tau, both unprimed and primed sites are phosphorylated by GSK3β, with Thr231 being the most notable primer epitope [26]. Although phosphorylation of S262 strongly reduced affinity for MT [27], phosphorylation of S214 [28] and T231 [29] primarily reduced Tau polymerization MTs. Ability [30] Phosphorylation of T231 not only regulates MT binding, but is also important for the role of Tau in disease [31] because it separates tau from MTS, which may Interaction with another cell partner [32]. Several kinases can phosphorylate Tau at T231, including glycogen synthase kinase 3β (GSK3β), one of the most important kinases involved in disease processes [33]. After initiation of phosphorylation at S235, GSK3β phosphorylates T231 more efficiently [34], even though this initiation of phosphorylation is not required [33]. Tau deposits isolated from Alzheimer’s disease patients typically contain phosphorylated T231 and S235, as well as phosphorylated S237 and S238 [35]. Furthermore, the unprimed site on GSK3β-R96A phosphorylated tau was more potent than wild-type GSK3β, clearly indicating the importance of priming site phosphorylation in regulating tau-microtubule interactions [36]. Following this preliminary study, it was demonstrated that GSK3β-induced tau phosphorylation of Thr231 plays a key role in reducing tau binding and stabilizing microtubules [37]. In transfected cells, tau with Thr231 mutated to Ala was still able to efficiently bind to microtubules after phosphorylation with GSK3β [37]. These studies clearly show that although GSK3β phosphorylates many sites on tau, not all sites have an effect on tau function.
Structure and Phosphorylation of Tau Protein
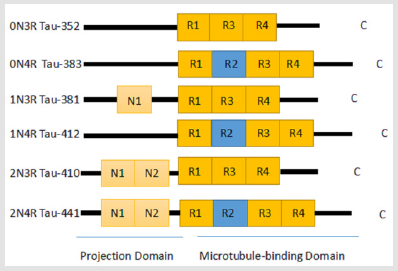
Figure 1: Different isomers of tau formed by alternative splicing of mRNA in the normal adult brain. The tau protein consists of two large regions: the projection domain (PD) and the microtubule-binding domain (MBD). The six unique isomers are mainly differentiated by the number of N-terminal insertion sequences (0N, 1N, or 2N) and the number of microtubule binding repeats (3R or 4R) that they contain. In the normal adult brain, the ratio of the 3R to 4R isomers is about 1:1.
The tau protein was identified in 1975 as a protein with the ability to induce microtubule formation [38,39]. It is the most widely occurring MAP in the normal brain and its primary function is to bind tubulin and promote its polymerization into microtubules [39]. It also combines with fully formed microtubules to maintain their stability [40], reduce the dissociation of tubulin molecules, and induce the formation of microtubule bundles. The tau gene is located on the long arm of chromosome 17 and has 79 phosphorylation sites that can be modified by serine/threonine protein kinases [38]. In fact, the polymerization and stabilization of microtubules are mainly determined by the state of tau phosphorylation. The phosphorylation of tau can be divided into two types depending on whether the modified residue is phosphorylated by a proline-directed kinase or a non-proline-directed kinase. Along the pathological course of many neurodegenerative diseases, tau protein is mainly (but not solely) phosphorylated by proline-directed protein kinases. In the central nervous system of a healthy human, alternate splicing of tau mRNA results in six different isoforms of the tau protein between 352–441 amino acids long with molecular weights of 48-67 kDa (Figure 1) [41-43]. The tau protein is subdivided into four regions: the acidic region at the N-terminal portion, the proline-rich region, the microtubule-binding domain, and the C-terminal region. Of the 85 putative phosphorylation sites in the tau protein, 45 sites are serines, 35 are threonines, and 5 are tyrosines [44-46].
Serine phosphorylation on the KXGS motif of the microtubule- binding domain reduces tau’s affinity for microtubules and thus prevents their binding [47-49]. The amount of tau protein phosphorylated at proline-rich sites like Thr-181, Ser-199, and Thr-231is higher in the brains of AD patients and these three phosphorylated forms of tau can therefore be used as biomarkers for AD [50-52]. Kinetic analysis showed that pseudophosphorylation increased the tau aggregation rate by increasing the filament nucleation rate. In addition, it increases the tendency to aggregate by stabilizing mature filaments to prevent depolymerization. The covalently bound phosphate is distributed within the tau microtubule- binding domain and adjacent to approximately 40 sites [45,53,54]. The occupancy of these sites may affect the tau aggregation in two ways. First, the occupancy of certain loci regulates the affinity of tau-tubulin [55], promoting an increase in the level of free cytoplasmic tau available for nucleation and supporting aggregation reactions [56-59]. Second, hyperphosphorylation directly increases the tendency of tau aggregation [60,61]. In addition, tau phosphorylation has been reported to reduce proteasome-mediated tau conversion in neuronal cell models [62]. Thus, the occupancy of certain tau phosphorylation sites can increase the free cytoplasmic tau concentration by a variety of mechanisms.
Tau phosphorylation and Neurological Diseases
Neurodegenerative diseases with abundant filamentous tau protein inclusion bodies are called tauopathies. Some neurodegenerative diseases differ from AD in that they lack the pathology of beta-amyloid plaques [63]. However, the tauopathies other than AD include chromosome 17-linked Parkinson’s disease with frontotemporal dementia, chronic traumatic encephalopathy, argicophilia granulosus, Progressive Supranuclear Palsy (PSP), corticobasal degeneration, globular glia tauopathy, and Pick’s disease. Due to the abnormal accumulation of phosphorylated tau protein in neuronal and glial cells in these neurodegenerative diseases, synaptic plasticity of hippocampal neurons can be affected, and memory function seriously disrupted [64]. It has been reported that changes in protein phosphorylation affect axonal transport in neurodegenerative disease models. For example, one study showed that as phosphorylation of neurofilament proteins and the microtubule-associated protein MAP1B increased, their respective axonal transport rates decreased [65].
In contrast, another study revealed that the enhanced phosphorylation level of tau increased the overall slow rate of tau protein transport in neurons and that the inhibition of tau phosphorylation by GSK-3 decreased its motility (Figure 2). Due to these and other similar findings, axonal transport defects have been regarded one of the contributing factors to neurodegenerative disease [66]. (Figure 2) Tau and other microtubule-associated proteins are phosphorylated by glycogen synthase kinase 3 (GSK-3), as well as cyclin- dependent kinases (like CDK5) and activator subunit p25, to form highly phosphorylated tau proteins. This highly phosphorylated form of the protein then dissociates into helical filaments that eventually form neurofibrillary tangles (NFTs) AD is acknowledged as the leading cause of dementia and is estimated to affect 47 million people worldwide [67]. The disease is primarily characterized by progressive cognitive and memory impairments. The neuropathological features of AD are (1) extensive cell death, (2) extracellular deposits of β-amyloid plaques (causing nephritis), and (3) synaptic aggregation of hyperphosphorylated tau protein also known as neurofibrillary tangles (NFTs) [68]. The analysis of the crystal structure of tau filaments in AD brains by Fitzpatrick et al. in 2017 showed that these pathological tau inclusions consist of paired helical filaments (PHFs) and straight filaments (SFs) [69- 71].
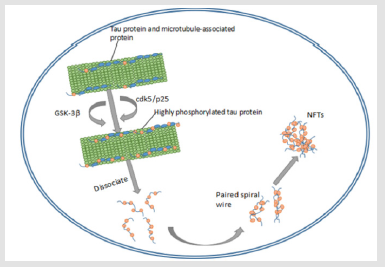
Figure 2: Tau and other microtubule-associated proteins are phosphorylated by glycogen synthase kinase 3 (GSK- 3), as well as cyclin-dependent kinases (like CDK5) and activator subunit p25, to form highly phosphorylated tau proteins. This highly phosphorylated form of the protein then dissociates into helical filaments that eventually form neurofibrillary tangles (NFTs).
Phosphorylation of tau enhances PHF formation. Phosphorylation can also be a physiologically feasible way to bring tau into a PHF-prone state. Phosphorylation can alter the conformation of tau, making it long and stiff [72]. Negative-stained electron microscopy showed that the core of the PHFs and SFs is composed of a double helix stack of C-shaped subunits [73] and successive steps along the β-strand of the protofilament are linked by helical symmetry. Moreover, the C-terminal region of tau is disordered, and it projects away from the core to form a fuzzy shell [74]. The protofilament cores of the PHFs and SFs are similar, indicating that they are ultrastructural polymorphs. The ultrastructural polymorphism between the PHF and SF is due to the difference in lateral contact between the two protofilaments. In the PHF, the two strands form exactly the same spiral symmetric structure, whereas in the SF, the protofilaments are asymmetric. In AD, tau is highly phosphorylated and many of the major kinases that phosphorylate the tau protein target glycogen synthase kinase-3 (GSK-3)-targeted tau phosphorylation sites [75]. Another of the major kinases responsible for tau hyperphosphorylation is cyclin-dependent kinase 5 (CDK5), a member of the serine/threonine kinase family of cyclin-dependent kinases.
Most AD neurons do not have normal microtubule structure but instead have pathological NFTs that are paired helical filaments of abnormal, hyperphosphorylated tau. Since tau pathology has been shown to be associated with neuronal loss, one of the treatment strategies targeting the molecular basis of AD includes inhibition of tau hyperphosphorylation [76]. To examine whether microtubule destruction induces tau phosphorylation, et al. co-expressed tau protein with stathmin, a 19 kDa phosphoprotein that depolymerizes microtubules, in COS-7 cells. Stathmin expression induced microtubule mutations and hyperphosphorylation of tau at Thr- 181, Ser-202, and Thr-205, indicating that microtubule disruption induces subsequent tau phosphorylation [77]. Frontotemporal lobar degeneration (FTLD) encompasses two clinical syndromes and three clinicopathological subtypes: the clinical syndromes are behavioral variant frontotemporal dementia and primary progressive aphasia, and the neuropathological subtypes are characterized by abnormal protein aggregation [64]. PSP is a rare, late-onset neurodegenerative disease whose clinical symptoms include early postural instability, vertical gaze palsy, and a later onset of dementia.
From the ultrastructural perspective, the NFT filaments present in PSP are straight and contain only the 4R isoform of the tau protein [78]. Animal models have revealed that mutations in the tau gene led to sprouting in dentate gyrus granule cells of hippocampal mossy fibers, and primary epilepsy is partially caused by mutations in the Tau protein gene. The S169L mutation of the presenilin 1 gene has also been found in patients with epileptic seizures and familial Alzheimer’s disease [79]. AD is the most common cause of dementia. It is a degenerative disease of the central nervous system and is mainly characterized by progressive cognitive impairment and memory impairment. The main pathological features of AD are senile plaques and neurofibrillary tangles. The core component of neurofibrillary tangles is the double-helical fibril formed by abnormally modified Tau protein [80]. Creutzfeldt-Jakob disease (CJD) is a rare and fatal human neurodegenerative disease that belongs to family of diseases known as transferable spongiform encephalopathies or prion diseases. The cerebrospinal fluid level in patients with CJD is significantly higher than that of AD patients and other dementia patients [81] (Table 1). As detailed above, it is clear that tau protein is closely associated with many diseases of the central nervous system and clarifying its mechanism of action can lead to new targets of treatment for tau protein-related diseases.
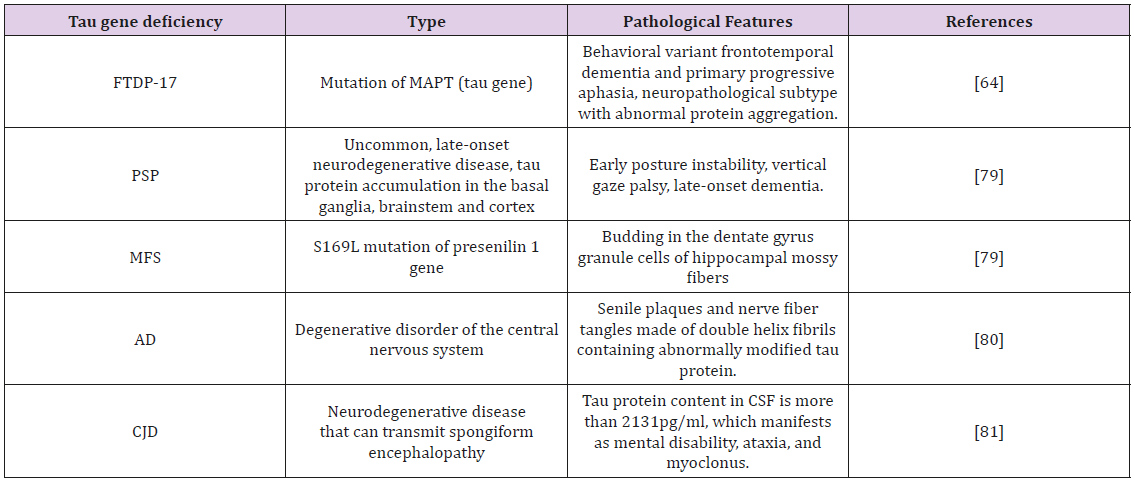
Table 1: Classification and characteristics of diseases caused by tau phosphorylation.
Phosphorylation Affects Axonal Transport and Degradation of the Tau protein
The phosphorylated form of the MAP tau accumulates in neurofibrillary tangles in Alzheimer’s disease. To investigate the effect of specific phosphorylated tau residues on protein function, expressed wild-type or phosphorylated tau protein in cultured cells. Their results showed that enhanced phosphorylation of tau decreased its microtubule binding and increased the number of moving tau particles without affecting axon transport kinetics. Conversely, decreasing tau protein phosphorylation increased the amount of tau protein bound to microtubules and inhibited axonal transport of tau. To determine whether the removal of tau protein resulted in an increase in phosphorylated tau, autophagy in neurons was inhibited. This resulted in a 3-fold increase in phosphorylated tau compared to wild-type tau and endogenous tau was not affected. In autophagy-deficient mouse embryonic fibroblasts, the proteasomal degradation of phosphorylated tau was also reduced compared with wild-type tau. These findings indicate that while both autophagy and proteasome pathways are involved in tau degradation, autophagy appears to be the main pathway for the removal of phosphorylated tau in neurons. Therefore, defective autophagy may contribute to the pathological accumulation of phosphorylated tau in neurodegenerative diseases [82].
Tauopathies are characterized by the presence of insoluble tau protein. The interaction of tau with microtubules is mainly achieved by the microtubule-binding domain located at the C-terminal of tau. This domain contains either three or four binding repeats (depending on alternative splicing of tenth exons), resulting in a 3R or 4R tau protein isomer, respectively. However, tau also interacts with components of the plasma membrane through its N-terminal projection domain [83]. While we know that phosphorylation of tau reduces its ability to bind and stabilize microtubules, we have recently found that the binding of tau to the plasma membrane is also regulated by phosphorylation [84]. It is well known that increased phosphorylation of tau reduces its affinity for microtubules, leading to instability of the neuronal cytoskeleton [85]. Phosphorylation specifically at Ser-262, Ser-293, Ser-324, and Ser-356, which are serines found in the KXGS sequences of R1, R2, R3, and R4 domains, respectively, have been shown to reduce the binding of tau to microtubules. Phosphorylation of tau in proline-rich regions surrounding Ser-202, Ser-235, Thr-231, and Ser-235 also contributes to the dissociation of tau from microtubules. However, phosphorylation in proline-rich regions alone is not enough to completely dissociate tau from microtubules [29]. GSK3β is a key protein in the insulin signaling pathway that phosphorylates several residues on tau [77,86]. The most favorable tau phosphorylation sites for GSK- 3β are Ser-396, Ser-400, and Ser-404 [87].
Phosphorylation of Ser-262 has been reported to result in reduced microtubule binding of tau [48,88]. However, phosphorylation of Ser-262 induced only about 40% of the microtubule binding activity [89], indicating that phosphorylation at other sites is necessary to completely inhibit its biological activity. The 21 phosphorylation sites in PHF-tau have been identified by reactivity with antibody and protein sequencing technologies at various phosphorylation sites. Among them, 10 sites are on the Ser / Thr- Pro motif and 11 are on the non-Ser / Thr-Pro motif [90,91]. Ser / Thr-Pro and non-Ser / Thr-Pro sites may be phosphorylated by proline- dependent protein kinase (PDPK) and non-PDPK, respectively. In the non-proline-directed phosphorylation site of PHF-tau, both Ser-208 and Ser-210 are in the SR-motif range.
In addition, in addition to the known GSK-3βphosphorylation site on tau, studies have identified a new phosphorylation site Thr- 175 and a non-prolineated phosphorylation site Ser-400. TTK is a non-proline-directed Ser / Thr kinase that has been purified from bovine brain [92]. It is the first tau kinase to phosphorylate Ser- 208 and Ser-210, both of which are PHE phosphate. Site. Thr-212 is a neighboring residue close to Ser-208 in tau and is known to be the phosphorylation site of GSK-3β [93]. Absorption tests by peptides pS208 and pS210 demonstrated the specificity of anti-pS208. Therefore, we can confirm that the phosphorylation site Ser-208 is a site separate from Thr-212. In addition to affecting its transport, tau phosphorylation also affects its ability to be degraded [94]. We studied the degradation of tau by the ubiquitin-proteasome system (UPS) and macroautophagy (autophagy) in the context of tau transport. While the UPS eliminates transient proteins by tagging them with chains of ubiquitin, autophagy removes long-lived structural proteins, as well as damaged or misfolded proteins [95]. Autophagy has also been shown to reduce both wild-type and modified tau proteins, including caspase-cleaved and C-terminally truncated species [96].
Tau protein hyperphosphorylation
Aberrant protein phosphorylation can lead to disease-related processes [97]. Accordingly, the abnormal phosphorylation of tau is observed in many neurodegenerative diseases. For example, histopathological investigations of AD showed extra-neuronal accumulation of β-amyloid peptide in plaques, neuronal aggregates of NFTs, and astrogliosis surrounding neurons [98]. Abnormal hyperphosphorylation of tau leads to aggregation, formation of NFTs, microtubule rupture, neuronal dysfunction, and death [99]. NFT consists of a pair of helical filaments (PHF), which in turn consists of a microtubule-associated protein tau in a hyperphosphorylated state [100]. In AD, the phosphorylation/dephosphorylation system appears to be greatly affected [101]. It has been shown that brain glucose uptake/metabolism in AD is impaired [102] and this damage has been suggested to be associated with abnormal hyperphosphorylation of tau. This finding implicates astrocytes as a key factor, especially because changes in glucose uptake and/or glutamate uptake (mediated by astrocytes) affect neuronal function and survival.
Through complex signal cascades, protein phosphorylation and dephosphorylation can regulate neuronal plasticity and neurotransmission, consequently impairing learning and memory. The signal cascade is precisely controlled by the dynamic reversible process of phosphorylation that is dependent on a precise balance between protein kinase and protein phosphatase activity. Human genome sequencing predicts the existence of more than 500 kinases and approximately 150 phosphatase genes. Protein kinases are subdivided into two families: serine/threonine kinases with 428 members; and tyrosine kinases with 90 members [103,104]. Protein phosphatases are categorized into three different families: protein tyrosine phosphatases (PTPs) [105], protein serine/threonine phosphatases (PSPs) [106], and dual-specificity protein phosphatases (tyrosine and serine/threonine). Of the known phosphatases, about 107 are PTPs and about 40 are PSPs [107].
Recent data show that the enzyme phosphatase family plays an indispensable role in controlling neuronal function [108]. The protein serine threonine phosphatase represents a highly conserved multigene family in evolution [109]. Based on sequence homology and biochemical properties, known phosphatases can be divided into four interrelated families. The three families of the protein serine threonine phosphatase types 1, 2A and 2B (PP1, PP2A and PP2B) have significant primary amino acid sequence homology, respectively. In contrast, phosphatase type 2C (PP2C) is more diverse. Among them, PP2A is a protein phosphatase that regulates the most phosphorylation of tau protein. Among the tau phosphatases identified in the human brain, PP2A accounts for more than 70% of tau dephosphorylation [110]. In the AD brain, PP2A activity was significantly reduced [111]. PP2A is a multimeric enzyme consisting of a catalytic subunit (C) and two regulatory subunits (A subunit or B subunit). The physiological form of PP2A is considered to be a heterogeneous composition composed of A and C subunits. Trimer. The major natural form of PP2A is a heterotrimer in which the core enzyme binds to one of several regulatory subunits expressed in a cell- and tissue-specific manner [112]. Another potential function of PP2A in the brain is to regulate phosphorylation of microtubule- associated protein (MAPS).
The activity of protein phosphatase (PP) 2A is downregulated and promotes hyperphosphorylation of tau in the brain of Alzheimer’s disease (AD). Studies have shown that calyculin A, a potent specific protein phosphatase (PP) 2A and PP1 inhibitor, is injected into both sides of the rat hippocampus, thereby replicating Alzheimer’s- like defects in the dephosphorylation system. It was found that rats injected with calyculin A found spatial memory retention damage in the Morris water maze test. At the same time, tau was hyperphosphorylated at the Ser396 / Ser404 (PHF-1) and Ser-262 / Ser-356 (12E8) sites, as determined by immunohistochemistry and Western blotting. This suggests that PP2A is involved in the in vivo regulation of tau phosphorylation and that down-regulation of this phosphatase will result in hyperphosphorylation of tau protein [113]. The hyperphosphorylation of the tau protein and the subsequent formation of NFTs are associated with abnormal activation of protein kinases [114].
In fact, studies have shown that the imbalance of kinase and phosphatase activity may play a causative role in the hyperphosphorylation of tau [102]. The proline-directed protein kinases that catalyze the phosphorylation of tau (such as GSK-3 and CDK5) predominantly do so at Ser-Pro and Thr-Pro sites on the tau protein, whereas the non-proline-directed protein kinases (such as protein kinase A, protein kinase C, calmodulin-dependent kinases, plasmin- dependent kinases, and glucocorticoid-dependent kinases) primarily phosphorylate serine or threonine residues and do not require proline guidance. It has been demonstrated at the cellular, brain, and animal levels that phosphatases play an important role in protein degradation in neurons in diseases such as AD. Studies have reported that inhibiting protein phosphatase activity induced tau hyperphosphorylation and aggregation [115].
Drugs that Affect Tau Phosphorylation Patterns
Nimodipine Attenuates Phosphorylation of Tau at Ser-396: Nimodipine is an L-type calcium channel antagonist that reduces excessive calcium influx in pathological conditions [116] and shows neuroprotective effects. Nimodipine treatment was initially used due to its ability to produce vasodilation in smooth muscle cells lined with blood vessels [117]. Chronic cerebral hypofusion (CCH) has been reported to promote hyperphosphorylation of the tau protein. It showed that nimodipine attenuated CCH-induced tau phosphorylation by up-regulating the expression of miR-132. In addition, nimodipine inhibited CCH-induced activation of GSK-3β and neuronal apoptosis. These findings support the role of nimodipine in inhibiting tau phosphorylation at Ser-396 via miR-132/GSK-3β and points to new potential drug target for the treatment of tauopathy in CCH by regulating the miR-132/GSK3β pathway [116].
Tamoxifen Inhibits CDK5 Kinase Activity and Regulates Tau Phosphorylation: CDK5 is a multifunctional enzyme that plays an important role in brain development. The catalytic subunit of this kinase does not have enzymatic activity as a monomer but is activated by binding to activation subunits p35 or p39. These activation subunits are structurally related to cyclins, activators of cell cycle CDKs, but do not show homology with cyclins at the amino acid level. In contrast to other CDKs, activation of CDK5 does not require phosphorylation of the activation loop. Studies have shown that neurotoxicity induces proteolytic cleavage of the p35 subunit by calcium-regulated calpains [68]. In vitro experiments have shown that this proteolytic conversion of p35 to p25 does not significantly alter the steady-state kinetics of tau phosphorylation by CDK5 [118]. The binding of CDK5 to p25, the N-terminally truncated proteolytic product, stabilizes CDK5 in the active dimer form and alters its substrate specificity.et al.identified tamoxifen from a large-scale bioluminescent resonance energy transfer (BRET)-based screen of small molecules that inhibit the interaction between CDK5 and p25. They showed that tamoxifen reduced tau phosphorylation by blocking the activation of CDK5 by p25 [118]. This finding paves the way for new therapies for tauopathies by harnessing the drug tamoxifen [118].
Rapamycin Reduces Tau Phosphorylation at Ser-214 by Modulating cAMP-Dependent kinases: Mammalian target of rapamycin (mTOR) is a highly evolutionarily conserved serine/threonine kinase. mTOR is involved in regulating many cellular processes such as autophagy, protein translation, ribosome biosynthesis, actin organization, mitochondrial oxygen consumption, proliferation, and differentiation [119]. It is worth noting that mTOR acts as a linker to protein kinase signals, receiving inputs from many upstream signaling pathways and delivering various downstream kinases such as cAMP-dependent protein kinases (e.g. PKA), GSK-3β, and mitogen-activated protein kinases [120]. Since all these kinases are tau-associated kinases, whether rapamycin can modulate tau phosphorylation by regulating these kinases remains to be determined. In human neuroblastoma SH-SY5Y cells, a cell model widely used for tau pathology studies, research indicated that rapamycin reduced the PKA-mediated phosphorylation of tau at Ser-214. Similar results were obtained in wild-type human embryonic kidney 293 (HEK293) cells that were stably transfected with the longest isoform of recombinant human tau (tau441; HEK293/tau441). Since Ser-214 is a site that blocks tau hyperphosphorylation [121], the inhibition of mTOR by rapamycin could indirectly prevent or reduce tau hyperphosphorylation.
Research has focused on rapamycin-induced enhancement of autophagy, as autophagy mediates massive degradation of cytoplasmic content and thus enhances the clearance of hyperphosphorylated tau [122]. It is also thought that rapamycin may inhibit the synthesis of the tau protein. However, since autophagyinduced by rapamycin gives priority to the reduction of excessive phosphorylated and insoluble tau and soluble tau is dispersed throughout the cell, it may not be easy to reduce tau levels by autophagic degradation, and showed that rapamycin improved memory deficits in 6-month-old 3xTg AD mice before accumulation of hyperphosphorylated and insoluble tau was observed [123]. Similarly, another study using an AD mouse model showed that the protective effect of rapamycin was apparent only before insoluble tau accumulated in these animals [124]. These studies suggest that the protective effects of rapamycin may not be limited to autophagic clearance of hyperphosphorylated and insoluble tau.
Conclusion
As a major MAP, tau protein plays an important role in neurodegenerative diseases. AD is pathologically identified by the presence of NFTs containing hyperphosphorylated tau protein. Glycosylation and ubiquitination also play a role in aberrant tau phosphorylation. Investigating the phosphorylation mechanisms of tau protein provides considerable insight into the progress of neurodegenerative diseases and can provide a reasonable basis for early disease treatment. Tubulin is a very unstable protein that easily loses GTP/ GDP exchange efficiency at 37°C without the presence of GTP and protein-stabilizing compounds with multiple hydroxyl groups. Thus, decreased tubulin turnover and/or the reduced expression of factors required for tubulin maintenance may decrease the number of microtubules or tubulin level in normal aging neurons. In addition, in autophagy-deficient mouse embryonic fibroblasts, but not in neurons, proteasomal degradation of phosphorylated tau is reduced compared to wild-type tau.
While autophagy and proteasome pathways are involved in tau degradation, autophagy appears to be the main pathway for the clearance of phosphorylated tau in neurons. The enhancement of autophagy pathways may have potential as a novel therapeutic strategy in AD and other neurodegenerative diseases, along with inhibiting in vivo signaling pathways that form hyperphosphorylated tau and proteins aggregates. Phosphorylation of the tau protein is regulated by inhibiting GSK-3β, CDK5, and activating PP2A acid esterase. In vitro cell culture studies have revealed that aniline, rhodanine, benzylhydrazide, amino pyridine, and other such compounds can inhibit the aggregation of tau. In recent years, a defect in kinase inactivation in old age has been suggested as a potential mechanism linking body temperature regulation and tau protein phosphorylation. This finding could provide a strategy to help the elderly improve their thermoregulatory mechanisms. It may also serve as a potential new AD treatment strategy.
Table Abbreviations
Frontotemporal dementia and tremor paralysis: FTDP-17, Microtubule Associated Protein Tau: MAPT, progressive supranuclear palsy: PSP, Marfan syndrome: MFS, Alzheimer’s disease: AD, Creutzfeldt-Jakob disease: CJD, Colony Stimulating Factor: CSF
Author Contributions
Conceptualization, Xu Han. and Keping Chen.; Validation, Keping Chen.; Formal Analysis, Xu Han; Investigation, Xu Han.; Writing – Original Draft Preparation, Xu Han; Writing – Review & Editing, Xu Han.; Visualization, Xu Han.; Supervision, Keping Chen; Project Administration, Keping Chen.; Funding Acquisition, Keping Chen.
Funding
This work was supported by the ational Natural Science Foundation of China [No. 31572467].
Acknowledgment
We would like to thank LetPub (www.LetPub.com) for providing linguistic assistance during the preparation of this manuscript.
Conflicts of Interest
The authors declare that they have no competing interests.
References
- Ramkumar A, Jong BY, Ori-Mc Kenney KM (2018) ReMAPping the microtubule landscape: How phosphorylation dictates the activities of microtubule-associated proteins. Developmental dynamics: an official publication of the American Association of Anatomists 247(1): 138-155.
- Buée L, Bussière T, Buéescherrer V, Delacourte A, Hof PR (2000) Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev 33(1): 95-130.
- Drubin DG, Kirschner MW (1986) Tau protein function in living cells. Journal of Cell Biology 103(6): 2739-2746.
- Köpke E, Tung YC, Shaikh S, Alonso AC, Iqbal K, et al. (1993) Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. Journal of Biological Chemistry 268(32): 24374-24384.
- Gong CX, Lidsky T, Wegiel J, Zuck L, Grundke-Iqbal I, et al. (2000) Phosphorylation of microtubule-associated protein tau is regulated by protein phosphatase 2A in mammalian brain. Implications for neurofibrillary degeneration in Alzheimer's disease. Journal of Biological Chemistry 275(8): 5535-5544.
- Spillantini MG, Goedert M (1998) Tau protein pathology in neurodegenerative diseases. Trends in Neurosciences 21(10): 428.
- Ballatore C, Lee VM, Trojanowski JQ (2007) Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nature Reviews Neuroscience 8(9): 663-672.
- Telzer BR, Haimo LT (1981) Decoration of spindle microtubules with Dynein: evidence for uniform polarity. J Cell Biol 89(2): 373-378.
- Stiess M, Maghelli N, Kapitein LC, Gomis Ruth S, Wilsch Brauninger M, et al. (2010) Axon Extension Occurs Independently of Centrosomal Microtubule Nucleation. Science 327(5966): 704-707.
- Baas PW, Rao AN, Matamoros AJ, Leo L (2016) Stability properties of neuronal microtubules. Cytoskeleton (Hoboken) 73(9): 442-460.
- Kirschner MW, Williams RC, Weingarten M, Gerhart JC (1974) Microtubules from mammalian brain: some properties of their depolymerization products and a proposed mechanism of assembly and disassembly. Proc Natl Acad Sci U S A 71(4): 1159-1163.
- Bieling P, Telley IA, Hentrich C, Piehler J, Surrey T (2010) Fluorescence microscopy assays on chemically functionalized surfaces for quantitative imaging of microtubule, motor, and+TIP dynamics. Methods Cell Biol 95: 555-580.
- Mitchison T, Kirschner M (1984) Dynamic instability of microtubule growth. Nature 312(5991): 237-242.
- Volle J, Brocard J, Saoud M, Goryfaure S, Brunelin J, et al. (2013) Reduced Expression of STOP/MAP6 in Mice Leads to Cognitive Deficits. Schizophrenia Bulletin 39(5): 969-978.
- Tymanskyj SR, Yang B, Falnikar A, Lepore AC, Ma L (2017) MAP7 Regulates Axon Collateral Branch Development in Dorsal Root Ganglia Neurons. Journal of Neuroscience the Official Journal of the Society for Neuroscience 37(6): 3260-3216.
- Ikegami K, Setou M (2010) Unique post-translational modifications in specialized microtubule architecture. Cell Struct Funct 35(1): 15-22.
- Miyasaka T, Sato S, Tatebayashi Y, Takashima A (2010) Microtubule destruction induces tau liberation and its subsequent phosphorylation. FEBS letters 584(14): 3227-3232.
- Hiller G, Weber K (1978) Radioimmunoassay for tubulin: a quantitative comparison of the tubulin content of different established tissue culture cells and tissues. Phosphorylation of the tau protein Cell 14(4): 795-804.
- Weisenberg RC (1972) Microtubule formation in vitro in solutions containing low calcium concentrations. Science (New York, NY) 177(4054): 1104-1105.
- Van Der Vaart B, Akhmanova A, Straube A (2009) Regulation of microtubule dynamic instability. Biochemical Society transactions 37(Pt 5): 1007-1013.
- Kirschner MW, Mitchison T (1986) Microtubule dynamics. Nature 324(6098): 621-621.
- Biernat J, Mandelkow EM (1999) The development of cell processes induced by tau protein requires phosphorylation of serine 262 and 356 in the repeat domain and is inhibited by phosphorylation in the proline-rich domains. Mol Biol Cell 10(3): 727-740.
- Keays DA (2007) Neuronal migration: unraveling the molecular pathway with humans, mice, and a fungus. Mamm Genome 18(6-7): 425-430.
- Martin N, Jaubert J, Gounon P, Salido E, Haase G, et al. (2002) A missense mutation in Tbce causes progressive motor neuronopathy in mice. Nature genetics 32(3): 443-447.
- Hollister R, West H, Mui S, Growdon JH, Petersen RC, et al. (2010) Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Annals of Neurology 41(1): 17-24.
- Zhou XZ, Kops O, Werner A, Lu PJ, Shen M, et al. (2000) Pin1-dependent prolyl isomerization regulates dephosphorylation of Cdc25C and tau proteins. Molecular cell 6(4): 873-883.
- Biernat Gustke, Drewes Mandelkow, Mandelkow (1993) Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: Distinction between PHF-like immunoreactivity and microtubule binding. Neuron 11(1): 153-163.
- Illenberger S, Zheng Fischhöfer Q, Preuss U, Stamer K, Baumann K, et al. (1998) The endogenous and cell cycle-dependent phosphorylation of tau protein in living cells: implications for Alzheimer's disease. Molecular Biology of the Cell 9(6): 1495.
- Amniai L, Barbier P, Sillen A, Wieruszeski JM, Peyrot V, et al. (2009) Alzheimer disease specific phosphoepitopes of Tau interfere with assembly of tubulin but not binding to microtubules. Faseb Journal 23 (4): 1146-1152.
- Sillen A, Barbier P, Landrieu I, Lefebvre S, Wieruszeski JM, et al. (2007) NMR investigation of the interaction between the neuronal protein tau and the microtubules. Biochemistry 46 (11): 3055-3064.
- Alonso AD, Di CJ, Li B, Corbo CP, Alaniz ME, et al. (2010) Phosphorylation of tau at Thr212, Thr231, and Ser262 combined causes neurodegeneration. Journal of Biological Chemistry 285(40): 30851.
- Frost B, Götz J, Feany MB (2015) Connecting the Dots Between Tau Dysfunction and Neurodegeneration. Trends in Cell Biology 25(1): 46-53.
- Lin YT, Cheng JT, Liang LC, Ko CY, Lo YK, et al. (2010) The binding and phosphorylation of Thr231 is critical for Tau’s hyperphosphorylation and functional regulation by glycogen synthase kinase 3β. Journal of Neurochemistry 103(2): 802-813.
- Li T, Hawkes C, Qureshi HY, Kar S, Paudel HK (2006) Cyclin-dependent protein kinase 5 primes microtubule-associated protein tau site-specifically for glycogen synthase kinase 3beta. Biochemistry 45 (10): 3134-3145.
- Hanger DP, Noble W (2011) Functional implications of glycogen synthase kinase-3-mediated tau phosphorylation. International Journal of Alzheimers Disease 2011: 352805.
- Cho JH, Johnson GV (2003) Glycogen synthase kinase 3beta phosphorylates tau at both primed and unprimed sites. Differential impact on microtubule binding. Journal of Biological Chemistry 278(1): 187-193.
- Cho JH, Johnson GV (2010) Primed phosphorylation of tau at Thr231 by glycogen synthase kinase 3beta (GSK3beta) plays a critical role in regulating tau's ability to bind and stabilize microtubules. Journal of Neurochemistry 88(2): 349-358.
- Cleveland DW, Hwo SY, Kirschner MW (1977) Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. Journal of Molecular Biology 116(2): 227-247.
- Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW (1975) A protein factor essential for microtubule assembly. Proceedings of the National Academy of Sciences of the United States of America 72 (5): 1858-1862.
- Kanai Y, Takemura R, Oshima T, Mori H, Ihara Y, et al. (1990) Expression of multiple tau isoforms and microtubule bundle formation in fibroblasts transfected with a single tau cDNA. Neuroscience Research Supplements 109(3): S79-S79.
- Gu Y, Oyama F, Ihara Y (1996) Tau is widely expressed in rat tissues. J Neurochem 67(3): 1235-1244.
- Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA (1989) Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J 8(2): 393-399.
- Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA (1989) Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron 3(4): 519-526.
- Sergeant N, Bretteville A, Hamdane M, Caillet-Boudin ML, Grognet P, et al. (2008) Biochemistry of Tau in Alzheimer's disease and related neurological disorders. Expert Rev Proteomics 5(2): 207-224.
- Hanger DP, Betts JC, Loviny TL, Blackstock WP, Anderton BH (1998) New phosphorylation sites identified in hyperphosphorylated tau (paired helical filament-tau) from Alzheimer's disease brain using nanoelectrospray mass spectrometry. J Neurochem 71(6): 2465-2476.
- Inoue H, Hiradate Y, Shirakata Y, Kanai K, Kosaka K, et al. (2014) Site-specific phosphorylation of Tau protein is associated with deacetylation of microtubules in mouse spermatogenic cells during meiosis. FEBS letters 588(11): 2003-2008.
- Sengupta A, Kabat J, Novak M, Wu Q, Grundke Iqbal I, et al. (1998) Phosphorylation of tau at both Thr 231 and Ser 262 is required for maximal inhibition of its binding to microtubules. Arch Biochem Biophys 357 (2): 299-309.
- Drewes G, Trinczek B, Illenberger S, Biernat J, Schmitt Ulms G, et al. (1995) Microtubule-associated protein/microtubule affinity-regulating kinase (p110mark). A novel protein kinase that regulates tau-microtubule interactions and dynamic instability by phosphorylation at the Alzheimer-specific site serine 262. J Biol Chem 270(13): 7679-7688.
- Dickey CA, Kamal A, Lundgren K, Klosak N, Bailey RM, et al. (2007) The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J Clin Invest 117(3): 648-658.
- Hampel H, Buerger K, Zinkowski R, Teipel SJ, Goernitz A, et al. (2004) Measurement of phosphorylated tau epitopes in the differential diagnosis of Alzheimer disease: a comparative cerebrospinal fluid study. Arch Gen Psychiatry 61(1): 95-102.
- Urakami K, Ishiguro K, Ohno H, Hampel H, Buerger K, et al. (2001) Large-scale, multicenter study of cerebrospinal fluid tau protein phosphorylated at serine 199 for the antemortem diagnosis of Alzheimer's disease. Ann Neurol 50(2): 150-156.
- Ishiguro K, Ohno H, Arai H, Yamaguchi H, Urakami K, et al. (1999) Phosphorylated tau in human cerebrospinal fluid is a diagnostic marker for Alzheimer's disease. Neurosci Lett 270(2): 91-94.
- Morishima Kawashima M, Hasegawa M, Takio K, Suzuki M, Yoshida H, et al. (1995) Hyperphosphorylation of tau in PHF. Neurobiol Aging 16(3): 365-380.
- Hanger DP, Byers HL, Wray S, Leung KY, Saxton MJ, et al. (2007) Novel Phosphorylation Sites in Tau from Alzheimer Brain Support a Role for Casein Kinase 1 in Disease Pathogenesis. Journal of Biological Chemistry 282(32): 23645.
- Ravier P, Luu GT, Jabloun M, Buttelli O (1984) The purification of tau protein and the occurrence of two phosphorylation states of tau in brain. Journal of Biological Chemistry 259(19): 12241-12245.
- Li G, Yin H, Kuret J (2004) Casein kinase 1 delta phosphorylates tau and disrupts its binding to microtubules. J Biol Chem 279(16): 15938-15945.
- Patrick GN, Zukerberg L, Nikolic M, De la Monte S, Dikkes P, et al. (1999) Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402(6762): 615-622.
- Biernat J, Gustke N, Drewes G, Mandelkow EM, Mandelkow E (1993) Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding. Neuron 11(1): 153-163.
- Bramblett GT, Goedert M, Jakes R, Merrick SE, Trojanowski JQ, et al. (1993) Abnormal tau phosphorylation at Ser396 in Alzheimer's disease recapitulates development and contributes to reduced microtubule binding. Neuron 10(6): 1089-1099.
- Alonso A, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K (2001) Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci U S A 98(12): 6923-6928.
- Alonso AD, Grundke Iqbal I, Barra HS, Iqbal K (1997) Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc Natl Acad Sci U S A 94(1): 298-303.
- Poppek D, Keck S, Ermak G, Jung T, Stolzing A, et al. (2006) Phosphorylation inhibits turnover of the tau protein by the proteasome: influence of RCAN1 and oxidative stress. Biochemical Journal 400(3): 511.
- Crowther RA, Goedert M (2000) Abnormal tau-containing filaments in neurodegenerative diseases. Journal of Structural Biology 130(2-3): 271-279.
- Combs B, Gamblin TC (2012) FTDP-17 tau mutations induce distinct effects on aggregation and microtubule interactions. Biochemistry 51(43): 8597-8607.
- Falzone TL, Gunawardena S, Mccleary D, Reis GF, Goldstein LSB (2010) Kinesin-1 transport reductions enhance human tau hyperphosphorylation, aggregation and neurodegeneration in animal models of tauopathies. Human Molecular Genetics 19(22): 4399-4408.
- Johnson GV (2006) Tau phosphorylation and proteolysis: insights and perspectives. Journal of Alzheimers Disease 9 (3 Suppl): 243-250.
- Winblad B, Amouyel P, Andrieu S, Ballard C, Brayne C, et al. (2016) Defeating Alzheimer's disease and other dementias: a priority for European science and society. Lancet Neurology 15(5): 455.
- Corbel C, Bing Z, Parc AL, Baratte B, Colas P, et al. (2015) Tamoxifen Inhibits CDK5 Kinase Activity by Interacting with p35/p25 and Modulates the Pattern of Tau Phosphorylation. Chemistry & Biology 22(4): 472-482.
- Yagishita S, Itoh Y, Nan W, Amano N (1981) Reappraisal of the fine structure of Alzheimer's neurofibrillary tangles. Acta Neuropathol 54(3): 239-246.
- Terry RD (1963) The Fine Structure of Neurofibrillary Tangles in Alzheimer's Disease. J Neuropathol Exp Neurol 22: 629-642.
- Kidd M (1963) Paired helical filaments in electron microscopy of Alzheimer's disease. Nature 197: 192-193.
- Hagestedt T, Lichtenberg B, Wille H, Mandelkow EM, Mandelkow E (1989) Tau protein becomes long and stiff upon phosphorylation: correlation between paracrystalline structure and degree of phosphorylation. Journal of Cell Biology 109 (4): 1643-1651.
- Crowther RA (1991) Straight and paired helical filaments in Alzheimer disease have a common structural unit. Proc Natl Acad Sci U S A 88 (6): 2288-2292.
- Wischik CM, Novak M, Edwards PC, Klug A, Tichelaar W, et al. (1988) Structural Characterization of the Core of the Paired Helical Filament of Alzheimer Disease. Proceedings of the National Academy of Sciences of the United States of America 85 (13): 4884-4888.
- Ackerley S, Thornhill P, Grierson AJ, Brownlees J, Anderton BH, et al. (2003) Neurofilament heavy chain side arm phosphorylation regulates axonal transport of neurofilaments. Journal of Cell Biology 161 (3): 489-495.
- Sundaram JR, Poore CP, Sulaimee NH, Pareek T, Asad AB, et al. (2013) Specific inhibition of p25/Cdk5 activity by the Cdk5 inhibitory peptide reduces neurodegeneration in vivo. Journal of Neuroscience 33 (1): 334-343.
- Liu K, Liu Y, Li L, Qin P, Iqbal J, et al. (2016) Glycation alter the process of Tau phosphorylation to change Tau isoforms aggregation property. Biochim Biophys Acta 1862 (2): 192-201.
- Moorhead GB, Trinkle-Mulcahy L, Nimick M, Wever VD, Campbell DG, et al. (2008) Displacement affinity chromatography of protein phosphatase one (PP1) complexes. BMC Biochemistry,9,1(2008-11-10) 9 (1): 28-28.
- Gold M, Lorenzl S, Stewart AJ, Morimoto BH, Williams DR, et al. (2012) Critical appraisal of the role of davunetide in the treatment of progressive supranuclear palsy. Neuropsychiatric Disease & Treatment 8 (default): 85-93.
- Arriagada PV, Growdon JH, Hedleywhyte ET, Hyman BT (1992) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology 42 (3 Pt 1): 631-639.
- Tagliapietra M, Zanusso G, Fiorini M, Bonetto N, Zarantonello G, et al. (2013) Accuracy of diagnostic criteria for sporadic creutzfeldt-jakob disease among rapidly progressive dementia. Journal of Alzheimers Disease Jad 34 (1): 231-238.
- Teresa RM, Inmaculada CI, Wendy N, Fanon N, Anderton BH, et al. (2013) Tau phosphorylation affects its axonal transport and degradation. Neurobiology of Aging 34 (9): 2146-2157.
- Pooler AM, Usardi A, Evans CJ, Philpott KL, Noble W, et al. (2012) Dynamic association of tau with neuronal membranes is regulated by phosphorylation. Neurobiol Aging 33(2): 431.e27-38.
- Avila J, León-Espinosa G, García E, García-Escudero V, Hernández F, et al. (2012) Tau Phosphorylation by GSK3 in Different Conditions. International Journal of Alzheimers Disease 2012 (5): 578373.
- Martin L, Latypova X, Terro F (2011) Post-translational modifications of tau protein: Implications for Alzheimer's disease. Neurochemistry International 58 (4): 458-471.
- Sui D, Liu M, Kuo MH (2015) In vitro aggregation assays using hyperphosphorylated tau protein. J Vis Exp (95): e51537.
- Leroy A, Landrieu I, Huvent I, Legrand D, Codeville B, et al. (2010) Spectroscopic Studies of GSK3β Phosphorylation of the Neuronal Tau Protein and Its Interaction with the N-terminal Domain of Apolipoprotein E. Journal of Biological Chemistry 285 (43): 33435-33444.
- Biernat J, Mandelkow EM, Schröter C, Lichtenberg-Kraag B, Steiner B, et al.(1992) The switch of tau protein to an Alzheimer-like state includes the phosphorylation of two serine-proline motifs upstream of the microtubule binding region. EMBO J 11 (4): 1593-1597.
- Singh TJ, Wang JZ, Novak M, Kontzekova E, Grundkeiqbal I, et al. (1996) Calcium/calmodulin-dependent protein kinase II phosphorylates tau at Ser-262 but only partially inhibits its binding to microtubules. Febs Letters 387 (3): 145-148.
- Iqbal K, Grundke Iqbal I (1995) Alzheimer abnormally phosphorylated tau is more hyperphosphorylated than the fetal tau and causes the disruption of microtubules. Neurobiology of Aging 16(3): 375-379.
- Morishima Kawashima M, Hasegawa M, Takio K, Suzuki M, Yoshida H, et al. (1995) Proline-directed and non-proline-directed phosphorylation of PHF-tau. Journal of Biological Chemistry 270(2): 823-829.
- Takahashi M, Tomizawa K, Sato K, Ohtake A, Omori A (1995) A novel tau-tubulin kinase from bovine brain. Febs Letters 372(1): 59-64.
- Tomizawa K, Omori A, Ohtake A, Sato K, Takahashi M (2001) Tau-tubulin kinase phosphorylates tau at Ser-208 and Ser-210, sites found in paired helical filament-tau. Febs Letters 492(3): 221-227.
- Shacka JJ, Roth KA, Zhang J (2008) The autophagy-lysosomal degradation pathway: role in neurodegenerative disease and therapy. Frontiers in Bioscience A Journal & Virtual Library 13: 718-736.
- Wang Y, Krüger U, Mandelkow E, Mandelkow EM (2010) Generation of Tau Aggregates and Clearance by Autophagy in an Inducible Cell Model of Tauopathy. Neurodegenerative Diseases 7(1-3): 103-107.
- Raskin J, Cummings J, Hardy J, Schuh K, Dean RA (2015) Neurobiology of Alzheimer’s Disease: Integrated Molecular, Physiological, Anatomical, Biomarker, and Cognitive Dimensions. Current Alzheimer Research 12(8): 712-722.
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S (2002) The protein kinase complement of the human genome. Science 298(5600): 1912-1934.
- Li B, Chohan MO, Grundke Iqbal I, Iqbal K (2007) Disruption of microtubule network by Alzheimer abnormally hyperphosphorylated tau. Acta Neuropathologica 113(5): 501-511.
- Cunnane S, Nugent S, Roy M, Courchesne Loyer A, Croteau E, et al. (2011) Brain fuel metabolism, aging, and Alzheimer’s disease. Nutrition 27 (1): 3-20.
- Lhara Y, Kondo J (1989) Polypeptide Composition of Paired Helical Filaments. Annals of Medicine 21(2): 121-125.
- Saitoh T, Masliah E, Jin LW, Cole GM, Wieloch T, et al. (1991) Protein kinases and phosphorylation in neurologic disorders and cell death. Laboratory Investigation 64 (5): 596-616.
- Gong CX, Liu F, Grundke Iqbal I, Iqbal K (2006) Dysregulation of protein phosphorylation/dephosphorylation in Alzheimer's disease: a therapeutic target. J Biomed Biotechnol 2006(3): 31825.
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378(6559): 785-789.
- Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, et al. (2004) Protein Tyrosine Phosphatases in the Human Genome. Cell 117(6): 699-711.
- Barford D (2003) The Structure and Topology of Protein Serine/Threonine Phosphatases. Elsevier Inc.
- Ceulemans H, Bollen M (2004) Functional diversity of protein phosphatase-1, a cellular economizer and reset button. Physiological Reviews 84(1): 1-39.
- Hernandez P, Lee GM, Maccioni R (2009) Tau phosphorylation by cdk5 and Fyn in response to amyloid peptide Abeta (25-35): involvement of lipid rafts. Journal of Alzheimers Disease 16(1): 149-156.
- Nairn AC, Shenolikar S (1992) The role of protein phosphatases in synaptic transmission, plasticity and neuronal development. Current Opinion in Neurobiology 2(3): 296-301.
- Depaoliroach AA, Park IK, Cerovsky V, Csortos C, Durbin SD, et al. (1994) Serine/threonine protein phosphatases in the control of cell function. Advances in Enzyme Regulation 34: 199-224.
- Liu F, Grundkeiqbal I, Iqbal K, Gong CX (2010) Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. European Journal of Neuroscience 22(8): 1942-1950.
- Gong CX, Shaikh S, Wang JZ, Zaidi T, Grundkeiqbal I, et al. (1995) Phosphatase activity toward abnormally phosphorylated tau: decrease in Alzheimer disease brain. Journal of Neurochemistry 65(2): 732-738.
- Wera S, Hemmings BA (1995) Serine/threonine protein phosphatases. Biochemical Journal 311 ( Pt 1): 17-29.
- Sun L, Liu SY, Zhou XW, Wang XC, Liu R, et al. (2003) Inhibition of protein phosphatase 2A- and protein phosphatase 1-induced tau hyperphosphorylation and impairment of spatial memory retention in rats. Neuroscience 118(4): 1175-1182.
- Tian Q, Wang J (2002) Role of Serine/Threonine Protein Phosphatase in Alzheimer’s Disease. Neurosignals 11(5): 262-269.
- Kumar S, Wirths O, Stüber K, Wunderlich P, Koch P, et al. (2016) Phosphorylation of the amyloid β-peptide at Ser26 stabilizes oligomeric assembly and increases neurotoxicity. Acta Neuropathol 131(4): 525-537.
- Tan Z, Chen Y, Xie W, Liu X, Zhu Y, et al. (2017) Nimodipine attenuates tau phosphorylation at Ser396 via miR-132/GSK-3β pathway in chronic cerebral hypoperfusion rats. European Journal of Pharmacology 819: 1-8.
- Brooks SP, Croft AP, Norman G, Shaw SG, Little HJ (2008) Nimodipine prior to alcohol withdrawal prevents memory deficits during the abstinence phase. Neuroscience 157(2): 376-384.
- Feldman HH, Haas M, Gandy S, Schoepp DD, Cross AJ, et al. (2014) Alzheimer's disease research and development: a call for a new research roadmap. Annals of the New York Academy of Sciences 1313: 1-16.
- Liu Y, Su Y, Wang J, Sun S, Wang T, et al. (2013) Rapamycin decreases tau phosphorylation at Ser214 through regulation of cAMP-dependent kinase. Neurochemistry International 62(4): 458-467.
- Li X, Han X, Llano J, Bole M, Zhou X, et al. (2011) Mammalian Target of Rapamycin Inhibition in Macrophages of Asymptomatic HIV+ Persons Reverses the Decrease in TLR-4-Mediated TNF-α Release through Prolongation of MAPK Pathway Activation. Journal of Immunology 187(11): 6052-6058.
- Liu S, Zhang J, Li H, Fang Z, Wang Q, et al. (2004) Tau becomes a more favorable substrate for GSK-3 when it is prephosphorylated by PKA in rat brain. J Biol Chem 279(48): 50078-50088.
- Caccamo A, Oddo S (2012) Inducing Autophagy by Rapamycin Before, but Not After, the Formation of Plaques and Tangles Ameliorates Cognitive Deficits. Alzheimers & Dementia the Journal of the Alzheimers Association 8(4): P201-201.
- Caccamo A, Majumder S, Richardson A, Strong R, Oddo S (2010) Molecular Interplay between Mammalian Target of Rapamycin (mTOR), Amyloid-β, and Tau: Effects on Cognitive Impairments. Journal of Biological Chemistry 285(17): 13107-13120.
- Nemoto T, Yanagita T, Satoh S, Maruta T, Kanai T, et al. (2011) Insulin-induced neurite-like process outgrowth: Acceleration of tau protein synthesis via a phosphoinositide 3-kinase∼mammalian target of rapamycin pathway. Neurochemistry International 59(6): 880-888.
