 Short Communication
Short CommunicationAbstract
Over the past decade, microbiologist have moved from testing individual antibiotic resistance genes (ARGs) to next-generation sequencing in which all known resistance genes within individual sample to detecting. This is providing a large amount of data on variation and relative number of ARGs in the total number of bacteria. However, it is necessary to analyze in the context of microbiome background to use this data in treatment or risk to the patient. With the help of a quantitative PCR ARG chip and 16S rRNA amplicon sequencing, with the use of genomic tools we have sought to identify ARG-like bacteria in healthy adult fecal samples. The 42 ARGs was detected, 12 fitted in ResCon1 category of ARG which is cphA, bacA, cfxA, blaTEM, aadE, aphA1, sul3, aphA3, aacA/aphd, catA1, vanC and aphA3. So, we describe these 12 genes as basic resistome of the fecal microbiome of this person and the remaining 30 ARGs as descriptors of the microbial community within the fecal microbiome. Dominant phyla and genera coincide with the previously identified maximum abundance samples in the fecal of healthy people. Most of the ARGs detected is associated with specific bacterial classification which confirmed the microbiome analysis. We admit the limitation of the data in the preference of data related to a limited sample set. However, the principle of the combination of qPCR and microbiome analysis is proof helps to identify the ARG Association with a taxa.
Short Communication
In order to minimize the burden of antibiotic resistance and identification of areas is the greatest risk to human health, to understand how genes of antibiotic resistance selection and spread can be found in complex bacterial system such as human intestinal microbiome. Not yet consensus on ARGs choice and bacterial changes required for the spread of ARGs and an increase in the number of ARG Complex bacterial population [1,2]. For understanding the effect of antibiotics on the mixed population complex such as gut microbiome we must first identify and understand the background or initial genetic resistance genes and internal resistant mechanisms present in the human gut bacteria [3- 5]. Then can we identify risks and potential conversion of ARG from intestinal microflora to pathogenic bacteria. If ARG is identified in the gut microbiome is present on the chromosome of an anaerobe [6]. It does not pose the same risk to the treatment of the patient as if the same gene is present in a highly mobile plasmid. Therefore, genes must be identified in connection with their bacterial or microbiomes. The aim of this study was to measure and identify the relative abundance of bacteria and ARG present in a fecal sample of a healthy adult using molecular biology tools [7]. As we move more towards genomic analysis of Bacteria and ARG, We need to develop the guidelines to do this we have to understand that which bacteria and ARG are present in healthy people and then what make a risk to the patient treatment [8].
Materials and Methods
Sample Preparation and DNA Extraction
Fecal samples were collected from a healthy person who haven’t been taken any antibiotics in the last two years. It’s immediately Homogenized, and 0.8 g are added directly to Fast DNA spin kit for soil DNA isolation kit in single step of the DNA separation. DNA is extracted using a kit protocol. The study was 16S rRNA amplicon sequencing V3 and V4 regions of the 16S rRNA gene are amplified and sequenced with Illumine Miseq [9-13]. 2⨯150 paired end configurations used for sequencing. Image analysis and base calls were processed using MiSeq control software. Data quality checking, trimmings and analysis were done through the Illumine Base Space software [14]. The sorted data were further processed by using Quantitative Insights into Microbial Ecology Versions (QIMME) 1.5.1 [15]. Shannon diversity, Chao 1 and collector curve were to conform uniformity of rRNA 16s results [16].
ARG Relative Abundance (QCPR)
DNA extracted from fecal samples was used to analyze Relative abundance of ARGs using Quantitative PCR Platform (qPCR) (Wafergen Smartchip) [17]. The analyzed samples include three biological replications per each sample and three technical replicates from each replicate. Samples were analyzed by relatively abundance of the 203 primers which has known MGE and ARG (Supplementary Table S1). Ct value of each primer pair is normalized with 16 rRNA gene value (deltaCt =CtARG-Ct16SrRNA). Results of a CT value of >28 has been deleted. The values of deltaCt and change in fold were calculated according to the relative gene expression data by use of quantitative PCR and deltadeltaCt methods [18]. Smart Chip has validated by Compared to metagenomic [19,20].
Results
ARG Resistome
Thirty-two different plus four mobile elements and two repressor genes were detected in the fecal sample (Table 1). By the use of the definitions of ARGs provided by Martinez et al. Exclude ampC genes, tetracycline resistance genes, efflux resistance genes and erythromycin resistance genes are not considered ARGs from an ecological perspective [9]. The tetracycline resistance genes and erythromycin resistance genes are identified frequently in common anaerobic chromosomes found microbiome of the human gut and it is not considered resistance to these bacteria. Their presence found the bacteria harboring these genes rather than a reservoir of resistance. Of the 32 detected ARGs, 11 belong to the category of ARGs ResCon1 defined by Martinez et al. [9]. These are the ARG bacA, cphA, sul3, aphA3, aphA1, aadE, aphD, vanC, cfxA and bla. So that’s why we describe these 10 genes as a major resistor in the fecal microbiome of this sampled person and the left 22 ARGs are descriptors for microbial community in the fecal microbiome.

Table 1: Antibiotic Resistance Genes abundance detected through Quantitative PCR chips.
Relative Abundance of Genes
The relative abundance of every ARGs was determined by Comparison with 16s rRNA genes abundance. This insured that variations in DNA quantities are not responsible for variations in the ARG abundance in the sample (Table 1). The highest relative abundance of genes Comprised of mobile genetic elements, tetracycline and erythromycin resistance genes is associated with anaerobe in the human gut microbiome Along with betalactamase cfxA gene Genes. The efflux genes tetR and aadE, an MDR resistance genes regulator bexA, VanC, Mobile genetics elements pni105MAP-F, a MLSB resistance genes msrC is the lowest relative abundance detected genes.
Bacterial Community Analysis using 16S rRNA Gene Amplicon Sequencing
Phyla which constitutes the taxa >1% in the 86302 OTU sequence through quality control filtering comprised Firmicutes (64%), Bacteroidetes (27%), Proteobacteria (2.4%), unclassified (1.6%) and Actinobacteria (1.3%). Dominant phyla are consistent with previous findings [16]. In these phyla the taxa were classified within 32 classes, with 6 Representing >1% relative abundance, Bacteroidia (64%), Clostridia (22%), Flavobacteria (7.3%), Unclassified (2.5%), Atinobacteria (1.6%) and Bacilli (1.2%). It Divided further into 68 orders, including Clostridiales (63%), Bacteroidales (21%), Flavobactriales (7.4), and Unclassified (2.4%) were representing >1%. Families in microbiome (n = 143) at >1% was presented Lachnospiraceae (31%), Ruminococcaceae (24%), Bacteroidaceae (12%), Flavobacteriaceae (7.5%), non-classified (4.4%), Clostridiaceae (3.7 %), Paraprevotellaceae (3.6%), Odoribacteraceae (2.5%), Porphyromonadaceae (2.1%) and Eubacteriaceae (1.5%).
A total of 262 different genera are represented in fecal microbiome and those with >1% of the relative abundance consist 19 different Genera (including non-classified), Represent 90% of the total composition of Microbiome (Figure 1). The dominant genera have previously agreed in the maximum abundance in feces samples from healthy people [10]. Although the correlation between bacterial polygenes and antibiotics resistome reported by Persson were using different habitats [11].
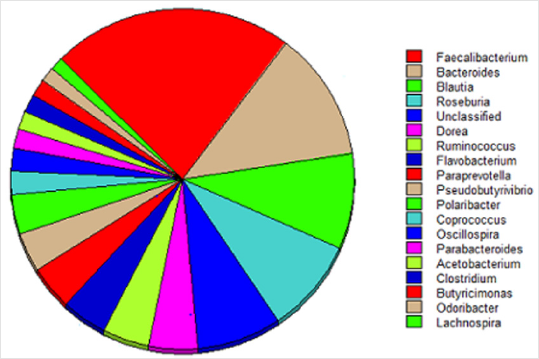
Figure 1: Spatial variation of the air pollution (NO2 and PM10) in the Cmpus.
Discussion
Bacteria is present in a mixed bacterial population like in human feces which contain ARGs on their chromosome or on mobile elements. Because of their role these bacteria are maintained in the fecal population, whether of the selective antibiotic pressure or due to ARG. With Next-generation sequencing progress research measuring the effects of antibiotics on complex bacteria population and its total antibiotic resistance, such as human gut microbiome [1-8]. However, to identify the changes in the population of the bacteria, we must first Identify the presence of ARG in natural fecal bacterial population, Same reduction Cost and capacity increase, NGS has become a potential tool to directly identify ARG and pathogens from the patient. In order to make such technology work properly, be sure to understand the difference between transportation and selection of bacterial species and ARG. The amid of this study was to integrate antibiotic resistance qPCR chips technology and NGS to explain the population of bacteria in human fecal and relative abundance of their resistome in healthy people under the lack of antibiotic pressure. The previous research on microbiom of the human gut alterate that antibiotics are highly variable. So far there are no conclusions defined on bacterial genera decreased or proliferate after specific antibiotic use [12].
There is no consensus on whether any microbiome changes arise or if interthematic variability has been effects of antibiotics administered. The first studies in the human gut microbiome tried to describe and find the basic set of bacterial taxa responsible for disease and health. However, this study in healthy individual reveals enormous differences in the composition of taxonomy of microbiome, which prevented discovery or finding core of microbiome. Most of the ARGs identified may be related to this that there is a specific bacterial classification. There is a high proportion of detected mobile genetic elements that indicates high genetic fluidity in the fecal microbiome and detect several ARG that are not related to intrinsic resistance to common gut microbiomes bacteria. The conclusion of this study is that, although a lot of ARG and its abundance can be detected using a DNA tool, we are sure so we put these sample in the context of bacterial composition of the sample, In one case a healthy human fecal to finding the Gene which show risk to the treatment of pathogenic infections. This study highlights the need to put DNA analysis into the list contains several ARGs that may be present in the chromosome of natural fecal microbiome. The conclusions cannot be extrapolated more due to a small sample size [13].
References
- Dethlefsen L, Huse S, Sogin ML, Relman DA (2008) The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol 6: e280.
- Sommer MO, Dantas G, Church (2009) Functional characterization of the antibiotic resistance reservoir in the human microflora. Science 325: 1128-1131.
- Jakobsson HE, Jernberg C, Andersson AF, Sjolund Karlsson M, Jansson JK (2010) Short-term antibiotic treatment has differing long-term impacts on the human throat and gut microbiome. PLoS One 5(3): e9836.
- Jernberg C, Lofmark S, Edlund C, Jansson JK (2010) Long-term impacts of antibiotic exposure on the human intestinal microbiota. Microbiology 156: 3216-3223.
- Claesson MJ, Cusack S, O’Sullivan O, Greene Diniz R, H de Weerd, et al. (2011) Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc Natl Acad Sci USA 108 Suppl 1: 4586-4591.
- Fouhy F, Guinane CM, Hussey S, Wall R, Ryan CA, et al. (2012) High-throughput sequencing reveals the incomplete, short-term recovery of infant gut microbiota following parenteral antibiotic treatment with ampicillin and gentamicin. Antimicrob. Agents Chemother 56(11): 5811-5820.
- Forslund K, Sunagawa S, Kultima JR, Mende DR, Arumugam M, et al. (2013) Country-specific antibiotic use practices impact the human gut resistome. Genome Res 23(7): 1163-1169.
- Pe´rez-Cobas AE, Gosalbes MJ, Friedrichs A, Knecht H, Artacho A, et al. (2013) Gut microbiota disturbance during antibiotic therapy: A multi-omic approach. Gut 62(1): 1591-1601.
- Martinez JL, TM Coque, Baquero F (2015) What is a resistance gene? Ranking risk in resistomes. Nat. Rev. Microbiol 13(2): 116-123.
- Lloyd-Price J, Abu-Ali G, Huttenhower C (2016) The healthy human microbiome. Genome Med 8: 51.
- Pehrsson EC, Tsukayama P, Patel S, Sosa-Soto G, Navarrete KM, et al. (2016) Interconnected microbiomes and resistomes in low-income human habitats. Nature 533(7602): 212-216.
- Turnbaugh PJ, RE Ley, Hamady M, CM Fraser Liggett, Rob Knight, et al. (2007) The human microbiome project. Nature 449: 804-810.
- Finegold SM, Sutter VL, Mathison GE (1983) Normal indigenous intestinal flora. In DJ Hentges (Eds.)., Human intestinal microflora in health and disease. Academic Press, Inc., New York, USA, p. 3-31.
- Illumina.
- Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Micro 73: 5261-5267.
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, et al. (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335-336.
- Parks DH, Tyson GW, Hugenholtz p, Beiko RG (2014) STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30(21): 3123-3124.
- Zhu YG, Johnson TA, Su JQ, Qiao M, Guo GX, et al. (2013) Diverse and abundant antibiotic resistance genes in Chinese swine farms. Proc. Natl Acad Sci USA 110(9): 3435-3440.
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)) method. Methods 25(4): 402-408.
- Stedtfeld RD, Stedtfeld TM, Fader KA, Williams MR, Bhaduri P, et al. (2017) TCDD influences reservoir of antibiotic resistance genes in murine gut microbiome. FEMS Microbiol Ecol 93(5): fix 058.
